
undefined
FATAL AUTOSOMAL DOMINANT DISORDER
GENE IN CHROMO 4p16.3
THE DISEASE IS ASSOCIATED WITH INCREASES LENGTH OF A CAG TRIPLET
[POLYGLUTAMINE] IN A GENE = HUNTINGTIN
DEGENERATION OF CHOLINERGIC AND GABAergic and ENKEPHALIN NEURONS IN THE BASAL GANGLIA AND THE CEREBRAL CORTEX [STRIATUM].Leading to increased activity of the inhibitory [GABAergic ] NEURONS FROM EXTERNAL GLOBUS PALLIDUS TO THE SUBTHALAMUS, WHICH CAUSES DECREASE IN THE GLUTAMATERGIC [EXCITATORY] NEURONAL ACTIVITY ON THE INTERNAL GLOBUS PALLIDUS. Result ia decreased inhibition on the thalamus, and the thalamus increases its excitatory [glutaminergic] effect on the cerebral cortex. Cause of the hyperkinesis.
PROGRESSIVE SELECTIVE NEURAL CELL DEATH ASSOCIATED WITH:
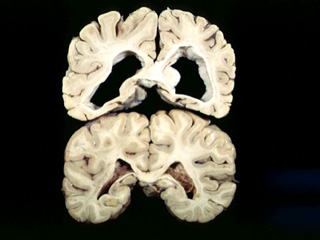
ATROPHY OF THE CAUDATE NUCLEUS.:
THERE IS A MARKED DEFICIENCY OF THE MITOCHONDRIAL RESPIRATORY CHAIN IN THE CAUDATE NUCLEUS.
CLASSIC SIGNS:
TYPICALLY THERE IS: PRODROMAL PHASE OF MILD PSYCHOTIC AND BEHAVIORAL SYMPTOMS
The persons begin with piano playing movements of the fingers or with slight facial twitching . The movements gradually becomes uncontrollable. The speech become incomprehensible and swallowing is difficult. The individuals lose the ability to communicate and are improperly nourished. Unable to stand or walk, and are confined to bed or to a wheelchair as the disease progresses. The ultimate result is death. |

II. DISEASES AFFECTING CORTEX & BASAL GANGLIA
C. Huntington's Disease
| Example of Huntington's disease pathology |
| Previous Section |
| Course Index |
| Section Index |
| Next
Section|
This pamphlet was written and published by the National Institute of Neurological Disorders and Stroke (NINDS), the United States' leading supporter of research on disorders of the brain and nervous system, including Huntington's disease. NINDS, one of the U.S. Government's 17 National Institutes of Health in Bethesda, Maryland, is part of the Public Health Service within the U.S. Department of Health and Human Services.
--Molecular Genetics
--The HD Gene and Its Product
--Cell Death in HD
--Animal Models in HD
--Fetal Tissue Research
--Clinical Studies
--Imaging
Introduction
In 1872, the American physician George Huntington wrote about an illness that he called "an heirloom from generations away back in the dim past." He was not the first to describe the disorder, which has been traced back to the Middle Ages at least. One of its earliest names was chorea,*which, as in "choreography," is the Greek word for dance. The term chorea describes how people affected with the disorder writhe, twist, and turn in a constant, uncontrollable dance-like motion. Later, other descriptive names evolved. "Hereditary chorea" emphasizes how the disease is passed from parent to child. "Chronic progressive chorea" stresses how symptoms of the disease worsen over time. Today, physicians commonly use the simple term Huntington's disease (HD) to describe this highly complex disorder that causes untold suffering for thousands of families.
In the United States alone, about 30,000 people have HD; estimates of its prevalenceare about 1 in every 10,000 persons. At least 150,000 others have a 50 percent risk of developing the disease and thousands more of their relatives live with the possibility that they, too, might develop HD.
Until recently, scientists understood very little about HD and could only watch as the disease continued to pass from generation to generation. Families saw the disease destroy their loved ones' ability to feel, think, and move. In the last several years, scientists working with support from the National Institute of Neurological Disorders and Stroke (NINDS) have made a significant number of breakthroughs in the area of HD research. With these advances, our understanding of the disease continues to improve.
This brochure presents information about HD, and about current research progress, to health professionals, scientists, caregivers, and, most importantly, to those already too familiar with the disorder: the many families who are affected by HD.
Return to table of contents
*see glossary
What Causes Huntingtons Disease?
HD results from genetically programmed degeneration of brain cells, called neurons, in certain areas of the brain. This degeneration causes uncontrolled movements, loss of intellectual faculties, and emotional disturbance. Specifically affected are cells of the basal ganglia, structures deep within the brain that have a number of important functions, including coordinating movement. Within the basal ganglia, HD especially targets neurons of the striatum, particularly those in the caudate nuclei and the pallidum. Also affected is the brain's outer surface, or cortex, which controls thought, perception, and memory.
Return to table of contents
How is HD Inherited?
HD is found in every country of the world. It is a familial disease, passed from parent to child through a mutation or misspelling in the normal gene.
A single abnormal gene, the basic biological unit of heredity, produces HD. Genes are composed of deoxyribonucleic acid (DNA), a molecule shaped like a spiral ladder. Each rung of this ladder is composed of two paired chemicals called bases. There are four types of bases--adenine, thymine, cytosine, and guanine--each abbreviated by the first letter of its name: A, T, C, and G. Certain bases always "pair" together, and different combinations of base pairs join to form coded messages. A gene is a long string of this DNA that is composed of various combinations of A, T, C, and G. These unique combinations determine the gene's function, much like letters join together to form words. Each person has about 100,000 genes--three billion base pairs of DNA or bits of information repeated in the nuclei of human cells--which determine individual characteristics or traits.
Genes are arranged in precise locations along 23 rod-like pairs of chromosomes. One chromosome from each pair comes from an individual's mother, the other from the father. Each half of a chromosome pair is similar to the other, except for one pair, which determines the sex of the individual. This pair has two x chromosomes in females and one x and one y chromosome in males. The gene that produces HD lies on chromosome 4, one of the 22 non-sex-linked, or "autosomal," pairs of chromosomes, placing men and women at equal risk of acquiring the disease.
The impact of a gene depends partly on whether it is dominantor recessive. If a gene is dominant, then only one of the paired chromosomes is required to produce its called-for effect. If the gene is recessive, both parents must provide chromosomal copies for the trait to be present. HD is called an autosomal dominant disorder because only one copy of the defective gene, inherited from one parent, is necessary to produce the disease.
The genetic defect responsible for HD is a small sequence of DNA on chromosome 4 in which several base pairs are repeated many, many times. The normal gene has three DNA bases, composed of the sequence CAG. In people with HD, the sequence abnormally repeats itself dozens of times. Over time--and with each successive generation--the number of CAG repeats may expand further.
Each parent has two copies of every chromosome but gives only one copy to each child. Each child of an HD parent has a 50-50 chance of inheriting the HD gene. If a child does not inherit the HD gene, he or she will not develop the disease and cannot pass it to subsequent generations. A person who inherits the HD gene, and survives long enough, will sooner or later develop the disease. In some families, all the children may inherit the HD gene; in others, none do. Whether one child inherits the gene has no bearing on whether others will or will not share the same fate.
A small number of cases of HD are sporadic, that is, they occur even though there is no family history of the disorder. These cases are thought to be caused by a new genetic mutationan alteration in the gene that occurs during sperm development and that brings the number of CAG repeats into the range that causes disease.
Return to table of contents
What are the Major Effects of the Disease?
Early signs of the disease vary greatly from person to person. A common observation is that the earlier the symptoms appear, the faster the disease progresses.
Family members may first notice that the individual experiences mood swings or becomes uncharacteristically irritable, apathetic, passive, depressed, or angry. These symptoms may lessen as the disease progresses or, in some individuals, may continue and include hostile outbursts or deep bouts of depression.
HD may affect the individual's judgment, memory, and other cognitive functions. Early signs might include having trouble driving, learning new things, remembering a fact, answering a question, or making a decision. Some may even display changes in handwriting. As the disease progresses, concentration on intellectual tasks becomes increasingly difficult.
In some individuals, the disease may begin with uncontrolled movements in the fingers, feet, face, or trunk. These movements--which are signs of chorea--often intensify when the person is anxious. HD can also begin with mild clumsiness or problems with balance. Other persons develop choreic movements later on as the disease progresses. They may stumble or appear uncoordinated. Chorea often creates serious problems with walking, increasing the likelihood of falls.
The disease can progress to the point where speech is slurred and vital functions, such as swallowing, eating, speaking, and especially walking, continue to decline. Some individuals are unable to recognize others. Many, however, remain aware of their environment and are able to express emotions.
Some physicians have employed a recently developed Unified HD Rating Scale, or UHDRS, to assess the clinical features, stages, and course of HD. In general, the duration of the illness ranges from 10 to 30 years. The most common causes of death are infection (most often pneumonia), injuries related to a fall, or other complications.
Return to table of contents
At What Age Does HD Appear?
The rate of disease progression and the age of onset vary from person to person. Adult-onset or classic HD, with its disabling, uncontrolled movements, most often begins during middle age. There are, however, other variations of HD distinguished not just by age of onset but by a distinct array of symptoms. For example, some persons develop the disease as adults, but without chorea. They may appear rigid and move very little, or not at all, a condition called akinesia. These individuals are said to have akinetic-rigid HD or the Westphal variant of HD.
Some individuals develop symptoms of HD when they are very young--before age 20. The terms early-onset HD or juvenile HD are often used to describe HD that appears in a young person. A common sign of HD in a younger individual is a rapid decline in school performance. Symptoms can also include subtle changes in handwriting and slight problems with movement, such as slowness, rigidity, tremor, and rapid muscular twitching, called myoclonus. Several of these symptoms are similar to those seen in Parkinson's disease, and they differ from the chorea seen in individuals who develop the disease as adults. People with juvenile HD may also have seizures and mental disabilities. As mentioned previously, the earlier the onset of HD, the faster the disease seems to progress. The disease progresses most rapidly in individuals with juvenile or early-onset HD, and death often follows within 10 years.
It appears that individuals with juvenile HD have usually inherited the disease from their fathers. These individuals also tend to have the largest number of CAG repeats. Scientists believe that the reason for this may be found in the process of sperm production. Unlike eggs, sperm are produced in the millions. Because DNA is copied millions of times during this process, scientists theorize that there is an increased possibility for genetic mistakes to occur. To verify that there was a link between the number of CAG repeats in the HD gene and the age of onset of the disease, scientists studied a young boy who developed HD at the age of two, one of the youngest and most severe cases ever recorded. They found that he had the largest number of CAG repeats of anyone they had studied so far--nearly 100. The boy's case was central to the identification of the HD gene and at the same time helped confirm that juvenile patients with HD have the longest segments of CAG repeats, the only proven correlation between repeat length and age at onset.
A few individuals develop HD after age 55. Diagnosis in these persons can be very difficult. The symptoms of HD may be masked by other health problems, or the person may not display the severity of symptoms seen in individuals with an earlier onset of HD. These individuals may also show signs of depression rather than anger or irritability, or they may retain sharp control over their intellectual functions, such as memory, reasoning, and problem-solving.
There is also a related complex called senile chorea. Some elderly individuals display the symptoms of HD, especially choreic movements, but have a normal gene and lack a family history of the disorder. Some scientists believe that a different gene mutation may account for this small number of cases. Others, however, believe senile chorea is a late-onset form of HD.
Return to table of contents
How is HD Diagnosed?
The great American folk singer and composer Woody Guthrie died on October 3, 1967, after suffering from HD for 13 years. He had been misdiagnosed, considered an alcoholic, and shuttled in and out of mental institutions and hospitals for years before being properly diagnosed. His case, sadly, is not extraordinary, although the diagnosis can be made easily by experienced neurologists.
The discovery of the HD gene in 1993 resulted in a direct genetic test to make or confirm a
diagnosis of HD in an individual who is exhibiting HD-like symptoms. Using a blood sample,
the genetic test analyzes DNA for the HD mutation by counting the number of repeats in
the HD gene region. Individuals who do not have HD usually have 28 or fewer CAG
repeats. Individuals with HD usually have 40 or more repeats. A small percentage of
individuals, however, have a number of repeats that fall within a borderline region (see
table 1).Table 1
No. of CAG repeats
Outcome
< 28
Normal range; individual will not develop HD
29-34
Individual will not develop HD but the next generation is at
risk
35-39
Some, but not all, individuals in this range will develop HD;
next generation is also at risk
> 40
Individual will develop HD
The physician will interview the individual intensively to obtain the medical history and rule out other conditions. He or she will perform a neurological examination including tests of the person's hearing, eye movements, strength, sensation, reflexes, balance, movement, and mental status, and will probably order a number of laboratory tests as well. Together, these tests form the neurological examination. In addition, the physician will ask about recent intellectual or emotional problems, which may be indications of HD.
In addition to direct testing, another tool used by physicians to diagnose HD is to take the family history, sometimes called a pedigree or genealogy. It is extremely important for family members to be candid and truthful with a doctor who is taking a family history.
People with HD commonly have impairments in the way the eye follows or fixes on a moving target. Abnormalities of eye movements vary from person to person and differ depending on the stage and duration of the illness.
The physician may ask the individual to undergo a brain imaging test. The computed tomography (CT) scanner provides an excellent image of brain structures with little if any discomfort. Those with HD may show shrinkage of some parts of the brain--particularly two areas known as the caudate nuclei and putamen--and enlargement of cavities within the brain called ventricles. These changes do not definitely indicate HD however, because they can also occur in other disorders. In addition, a person can have early symptoms of HD and still have a normal CT scan. When used in conjunction with a family history and record of clinical symptoms, however, CT can be an important diagnostic tool.
Other technologies for brain visualization, such as magnetic resonance imaging (MRI)and positron emission tomography (PET), are an important part of HD research efforts, but their usefulness to physicians trying to diagnose HD has not yet been established.
Return to table of contents
What is Presymptomatic Testing?
Presymptomatic testing is a method for identifying persons carrying the HD gene before symptoms appear. In the past, no laboratory test could positively identify people carrying the HD gene--or those fated to develop HD--before the onset of symptoms. That situation changed in 1983, when a team of scientists supported by the NINDS located the first genetic markerfor HD--the initial step in developing a laboratory test for the disease.
A marker is a piece of DNA that lies near a gene and is usually inherited with it. Discovery of the first HD marker allowed scientists to locate the HD gene on chromosome 4. The marker discovery quickly led to the development of a presymptomatic test for some individuals, but this test required blood or tissue samples from both affected and unaffected family members in order to identify markers unique to that particular family. For this reason, adopted individuals, orphans, and people who had few living family members were unable to use the test.
Discovery of the HD gene has led to a less expensive, scientifically simpler, and far more accurate presymptomatic test that is applicable to the majority of at-riskpeople. The new test uses CAG repeat length to detect the presence of the HD mutation in blood. This is discussed further in the next section.
In a small number of individuals with HD--1 to 3 percent--no family history of HD can be found. Some individuals may not be aware of their genetic legacy, or a family member may conceal a genetic disorder from fear of social stigma. A parent may not want to worry children, scare them, or deter them from marrying. In other cases, a family member may die of another cause before he or she begins to show signs of HD. Sometimes, the cause of death for a relative may not be known, or the family is not aware of a relative's death. Adopted children may not know their genetic heritage, or early symptoms in an individual may be too slight to attract attention. These are among the many complicating factors that reflect the complexity of diagnosing HD.
Return to table of contents
How is the Presymptomatic Test Conducted?
An individual who wishes to be tested should contact the nearest testing center. (A list of such centers can be obtained from the Huntington Disease Society of America at 1-800-345-HDSA.) The testing process should include several components. Most testing programs include a neurological examination, pretest counseling, and followup. The purpose of the neurological examination is to determine whether or not the person requesting testing is showing any clinical symptoms of HD. It is important to remember that if an individual is showing even slight symptoms of HD, he or she risks being diagnosed with the disease during the neurological examination, even before the genetic test. During pretest counseling, the individual will learn about HD, about his or her own level of risk, and about the testing procedure. The person will be told about the test's limitations, the accuracy of the test, and possible outcomes. He or she can then weigh the risks and benefits of testing and may even decide at that time against pursuing further testing.
If a person decides to be tested, a team of highly trained specialists will be involved, which may include neurologists, genetic counselors, social workers, psychiatrists, and psychologists. This team of professionals helps the at-risk person decide if testing is the right thing to do and carefully prepares the person for a negative, positive, or inconclusive test result.
Individuals who decide to continue the testing process should be accompanied to counseling sessions by a spouse, a friend, or a relative who is not at risk. Other interested family members may participate in the counseling sessions if the individual being tested so desires.
The genetic testing itself involves donating a small sample of blood that is screened in the laboratory for the presence or absence of the HD mutation. Testing may require a sample of DNA from a closely related affected relative, preferably a parent, for the purpose of confirming the diagnosis of HD in the family. This is especially important if the family history for HD is unclear or unusual in some way.
Results of the test should be given only in person and only to the individual being tested. Test results are confidential. Regardless of test results, followup is recommended.
In order to protect the interests of minors, including confidentiality, testing is not recommended for those under the age of 18 unless there is a compelling medical reason (for example, the child is exhibiting symptoms).
Testing of a fetus (prenatal testing) presents special challenges and risks; in fact some centers do not perform genetic testing on fetuses. Because a positive test result using direct genetic testing means the at-risk parent is also a gene carrier, at-risk individuals who are considering a pregnancy are advised to seek genetic counseling prior to conception.
Some at-risk parents may wish to know the risk to their fetus but not their own. In this situation, parents may opt for prenatal testing using linked DNA markers rather than direct gene testing. In this case, testing does not look for the HD gene itself but instead indicates whether or not the fetus has inherited a chromosome 4 from the affected grandparent or from the unaffected grandparent on the side of the family with HD. If the test shows that the fetus has inherited a chromosome 4 from the affected grandparent, the parents then learn that the fetus's risk is the same as the parent (50-50), but they learn nothing new about the parent's risk. If the test shows that the fetus has inherited a chromosome 4 from the unaffected grandparent, the risk to the fetus is very low (less than 1%) in most cases.
Another option open to parents is in vitro fertilization with preimplantation screening. In this procedure, embryos are screened to determine which ones carry the HD mutation. Embryos determined not to have the HD gene mutation are then implanted in the woman's uterus.
In terms of emotional and practical consequences, not only for the individual taking the test but for his or her entire family, testing is enormously complex and has been surrounded by considerable controversy. For example, people with a positive test result may risk losing health and life insurance, suffer loss of employment, and other liabilities. People undergoing testing may wish to cover the cost themselves, since coverage by an insurer may lead to loss of health insurance in the event of a positive result, although this may change in the future.
With the participation of health professionals and people from families with HD, scientists have developed testing guidelines. All individuals seeking a genetic test should obtain a copy of these guidelines, either from their testing center or from the organizations listed on the card in the back of this brochure. These organizations have information on sites that perform testing using the established procedures and they strongly recommend that individuals avoid testing that does not adhere to these guidelines.
Return to table of contents
How Does a Person Decide Whether to be Tested?
The anxiety that comes from living with a 50 percent risk for HD can be overwhelming. How does a young person make important choices about long-term education, marriage, and children? How do older parents of adult children cope with their fears about children and grandchildren? How do people come to terms with the ambiguity and uncertainty of living at risk?
Some individuals choose to undergo the test out of a desire for greater certainty about their genetic status. They believe the test will enable them to make more informed decisions about the future. Others choose not to take the test. They are at peace with being at risk and with all that that may entail. There is no right or wrong decision, as each choice is highly individual. The guidelines for genetic testing for HD, discussed in the previous section, were developed to help people with this life-changing choice.
Whatever the results of genetic testing, the at-risk individual and family members can expect powerful and complex emotional responses. The health and happiness of spouses, brothers and sisters, children, parents, and grandparents are affected by a positive test result, as are an individual's friends, work associates, neighbors, and others. Because receiving test results may prove to be devastating, testing guidelines call for continued counseling even after the test is complete and the results are known.
Return to table of contents
Is There a Treatment for HD?
Physicians may prescribe a number of medications to help control emotional and movement problems associated with HD. It is important to remember however, that while medicines may help keep these clinical symptoms under control, there is no treatment to stop or reverse the course of the disease.
Antipsychotic drugs, such as haloperidol, or other drugs, such as clonazepam, may help to alleviate choreic movements and may also be used to help control hallucinations, delusions, and violent outbursts. Antipsychotic drugs, however, are not prescribed for another form of muscle contraction associated with HD, called dystonia, and may in fact worsen the condition, causing stiffness and rigidity. These medications may also have severe side effects, including sedation, and for that reason should be used in the lowest possible doses.
For depression, physicians may prescribe fluoxetine, sertraline hydrochloride, nortriptyline, or other compounds. Tranquilizers can help control anxiety and lithium may be prescribed to combat pathological excitement and severe mood swings. Medications may also be needed to treat the severe obsessive-compulsive rituals of some individuals with HD.
Most drugs used to treat the symptoms of HD have side effects such as fatigue, restlessness, or hyperexcitability. Sometimes it may be difficult to tell if a particular symptom, such as apathy or incontinence, is a sign of the disease or a reaction to medication.
Return to table of contents
What Kind of Care Does the Individual with HD Need?
Although a psychologist or psychiatrist, a genetic counselor, and other specialists may be needed at different stages of the illness, usually the first step in diagnosis and in finding treatment is to see a neurologist. While the family doctor may be able to diagnose HD, and may continue to monitor the individuals status, it is better to consult with a neurologist about management of the varied symptoms.
Problems may arise when individuals try to express complex thoughts in words they can no longer pronounce intelligibly. It can be helpful to repeat words back to the person with HD so that he or she knows that some thoughts are understood. Sometimes people mistakenly assume that if individuals do not talk, they also do not understand. Never isolate individuals by not talking, and try to keep their environment as normal as possible. Speech therapy may improve the individuals ability to communicate.
It is extremely important for the person with HD to maintain physical fitness as much as his or her condition and the course of the disease allows. Individuals who exercise and keep active tend to do better than those who do not. A daily regimen of exercise can help the person feel better physically and mentally. Although their coordination may be poor, individuals should continue walking, with assistance if necessary. Those who want to walk independently should be allowed to do so as long as possible, and careful attention should be given to keeping their environment free of hard, sharp objects. This will help ensure maximal independence while minimizing the risk of injury from a fall. Individuals can also wear special padding during walks to help protect against injury from falls. Some people have found that small weights around the ankles can help stability. Wearing sturdy shoes that fit well can help too, especially shoes without laces that can be slipped on or off easily.
Impaired coordination may make it difficult for people with HD to feed themselves and to swallow. As the disease progresses, persons with HD may even choke. In helping individuals to eat, caregivers should allow plenty of time for meals. Food can be cut into small pieces, softened, or pureed to ease swallowing and prevent choking. While some foods may require the addition of thickeners, other foods may need to be thinned. Dairy products, in particular, tend to increase the secretion of mucus, which in turn increases the risk of choking. Some individuals may benefit from swallowing therapy, which is especially helpful if started before serious problems arise. Suction cups for plates, special tableware designed for people with disabilities, and plastic cups with tops can help prevent spilling. The individual's physician can offer additional advice about diet and about how to handle swallowing difficulties or gastrointestinal problems that might arise, such as incontinence or constipation.
Caregivers should pay attention to proper nutrition so that the individual with HD takes in enough calories to maintain his or her body weight. Sometimes people with HD, who may burn as many as 5,000 calories a day without gaining weight, require five meals a day to take in the necessary number of calories. Physicians may recommend vitamins or other nutritional supplements. In a long-term care institution, staff will need to assist with meals in order to ensure that the individual's special caloric and nutritional requirements are met. Some individuals and their families choose to use a feeding tube; others choose not to.
Individuals with HD are at special risk for dehydration and therefore require large quantities of fluids, especially during hot weather. Bendable straws can make drinking easier for the person. In some cases, water may have to be thickened with commercial additives to give it the consistency of syrup or honey.
Return to table of contents
What Community Resources are Available?
Individuals and families affected by HD can take steps to ensure that they receive the best advice and care possible. Physicians and state and local health service agencies can provide information on community resources and family support groups that may exist. Possible types of help include:
Legal and social aid. HD affects a person's capacity to reason, make judgments, and handle responsibilities. Individuals may need help with legal affairs. Wills and other important documents should be drawn up early to avoid legal problems when the person with HD may no longer be able to represent his or her own interests. Family members should also seek out assistance if they face discrimination regarding insurance, employment, or other matters.
Home care services. Caring for a person with HD at home can be exhausting, but part-time assistance with household chores or physical care of the individual can ease this burden. Domestic help, meal programs, nursing assistance, occupational therapy, or other home services may be available from federal, state, or local health service agencies.
Recreation and work centers. Many people with HD are eager and able to participate in activities outside the home. Therapeutic work and recreation centers give individuals an opportunity to pursue hobbies and interests and to meet new people. Participation in these programs, including occupational, music, and recreational therapy, can reduce the persons dependence on family members and provides home caregivers with a temporary, much needed break.
Group housing. A few communities have group housing facilities that are supervised by a resident attendant and that provide meals, housekeeping services, social activities, and local transportation services for residents. These living arrangements are particularly suited to the needs of individuals who are alone and who, although still independent and capable, risk injury when they undertake routine chores like cooking and cleaning.
Institutional care. The individuals physical and emotional demands on the family may eventually become overwhelming. While many families may prefer to keep relatives with HD at home whenever possible, a long-term care facility may prove to be best. To hospitalize or place a family member in a care facility is a difficult decision; professional counseling can help families with this.
Finding the proper facility can itself prove difficult. Organizations such as the Huntington's Disease Society of America (see Information Resources) may be able to refer the family to facilities that have met standards set for the care of individuals with HD. Very few of these exist however, and even fewer have experience with individuals with juvenile or early-onset HD who require special care because of their age and symptoms.
Return to table of contents
What Research is Being Done?
Although HD attracted considerable attention from scientists in the early 20th century, there was little sustained research on the disease until the late 1960s when the Committee to Combat Huntingtons Disease and the Huntingtons Chorea Foundation, later called the Hereditary Disease Foundation, first began to fund research and to campaign for federal funding. In 1977, Congress established the Commission for the Control of Huntington's Disease and Its Consequences, which made a series of important recommendations. Since then, Congress has provided consistent support for federal research, primarily through the National Institute of Neurological Disorders and Stroke, the governments lead agency for biomedical research on disorders of the brain and nervous system. The effort to combat HD proceeds along the following lines of inquiry, each providing important information about the disease:
Basic neurobiology. Now that the HD gene has been located, investigators in the field of neurobiologywhich encompasses the anatomy, physiology, and biochemistry of the nervous systemare continuing to study the HD gene with an eye toward understanding how it causes disease in the human body.
Clinical research. Neurologists, psychologists, psychiatrists, and other investigators are improving our understanding of the symptoms and progression of the disease in patients while attempting to develop new therapeutics.
Imaging. Scientific investigations using PET and other technologies are enabling scientists to see what the defective gene does to various structures in the brain and how it affects the body's chemistry and metabolism.
Animal models. Laboratory animals, such as mice, are being bred in the hope of duplicating the clinical features of HD and can soon be expected to help scientists learn more about the symptoms and progression of the disease.
Fetal tissue research. Investigators are implanting fetal tissue in rodents and nonhuman primates with the hope that success in this area will lead to understanding, restoring, or replacing functions typically lost by neuronal degeneration in individuals with HD.
These areas of research are slowly converging and, in the process, are yielding important clues about the gene's relentless destruction of mind and body. The NINDS supports much of this exciting work.
Molecular Genetics
For 10 years, scientists focused on a segment of chromosome 4 and, in 1993, finally isolated the HD gene. The process of isolating the responsible gene--motivated by the desire to find a cure--was more difficult than anticipated. Scientists now believe that identifying the location of the HD gene is the first step on the road to a cure.
Finding the HD gene involved an intense molecular genetics research effort with cooperating investigators from around the globe. In early 1993, the collaborating scientists announced they had isolated the unstable triplet repeat DNA sequence that has the HD gene. Investigators relied on the NINDS-supported Research Roster for Huntington's Disease, based at Indiana University in Indianapolis, to accomplish this work. First started in 1979, the roster contains data on many American families with HD, provides statistical and demographic data to scientists, and serves as a liaison between investigators and specific families. It provided the DNA from many families affected by HD to investigators involved in the search for the gene and was an important component in the identification of HD markers.
For several years, NINDS-supported investigators involved in the search for the HD gene made yearly visits to the largest known kindredwith HD--14,000 individuals--who live on Lake Maracaibo in Venezuela. The continuing trips enable scientists to study inheritance patterns of several interrelated families.
The HD Gene and Its Product
Although scientists know that certain brain cells die in HD, the cause of their death is still unknown. Recessive diseases are usually thought to result from a gene that fails to produce adequate amounts of a substance essential to normal function. This is known as a loss-of-function gene. Some dominantly inherited disorders, such as HD, are thought to involve a gene that actively interferes with the normal function of the cell. This is known as a gain-of-function gene.
How does the defective HD gene cause harm? The HD gene encodes a protein--which has been named huntingtin--the function of which is as yet unknown. The repeated CAG sequence in the gene causes an abnormal form of huntingtin to be made, in which the amino acid glutamine is repeated. It is the presence of this abnormal form, and not the absence of the normal form, that causes harm in HD. This explains why the disease is dominant and why two copies of the defective gene--one from both the mother and the father--do not cause a more serious case than inheritance from only one parent. With the HD gene isolated, NINDS-supported investigators are now turning their attention toward discovering the normal function of huntingtin and how the altered form causes harm. Scientists hope to reproduce, study, and correct these changes in animal models of the disease.
Huntingtin is found everywhere in the body but only outside the cells nucleus. Mice bred in the laboratory to produce no huntingtin fail to develop past a very early embryo stage and quickly die. Huntingtin, scientists now know, is necessary for life. Investigators hope to learn why the abnormal version of the protein damages only certain parts of the brain. One theory is that cells in these parts of the brain may be supersensitive to this abnormal protein.
Return to table of contents
Cell Death in HD
Although the precise cause of cell death in HD is not yet known, scientists are paying close attention to the process of genetically programmed cell death that occurs deep within the brains of individuals with HD. This process involves a complex series of interlinked events leading to cellular suicide. Related areas of investigation include:
Several HD studies are aimed at understanding losses of nerve cells and receptorsin HD. Neurons in the striatum are classified both by their size (large, medium, or small) and appearance (spiny or aspiny). Each type of neuron contains combinations of neurotransmitters. Scientists know that the destructive process of HD affects different subsets of neurons to varying degrees. The hallmark of HD, they are learning, is selective degeneration of medium-sized spiny neurons in the striatum. NINDS-supported studies also suggest that losses of certain types of neurons and receptors are responsible for different symptoms and stages of HD.
What do these changes look like? In spiny neurons, investigators have observed two types of changes, each affecting the nerve cells' dendrites. Dendrites, found on every nerve cell, extend out from the cell body and are responsible for receiving messages from other nerve cells. In the intermediate stages of HD, dendrites grow out of control. New, incomplete branches form and other branches become contorted. In advanced, severe stages of HD, degenerative changes cause sections of dendrites to swell, break off, or disappear altogether. Investigators believe that these alterations may be an attempt by the cell to rebuild nerve cell contacts lost early in the disease. As the new dendrites establish connections, however, they may in fact contribute to nerve cell death. Such studies give compelling, visible evidence of the progressive nature of HD and suggest that new experimental therapies must consider the state of cellular degeneration. Scientists do not yet know exactly how these changes affect subsets of nerve cells outside the striatum.
Return to table of contents
Animal Models for HD
As more is learned about cellular degeneration in HD, investigators hope to reproduce these changes in animal models and to find a way to correct or halt the process of nerve cell death. Such models serve the scientific community in general by providing a means to test the safety of new classes of drugs in nonhuman primates. NINDS-supported scientists are currently working to develop both nonhuman primate and mouse models to investigate nerve degeneration in HD and to study the effects of excitotoxicity on nerve cells in the brain.
Investigators are working to build genetic models of HD using transgenic mice. To do this, scientists transfer the altered human HD gene into mouse embryos so that the animals will develop the anatomical and biological characteristics of HD. This genetic model of mouse HD will enable in-depth study of the disease and testing of new therapeutic compounds.
Another idea is to insert into mice a section of DNA containing CAG repeats in the abnormal, disease gene range. This mouse equivalent of HD could allow scientists to explore the basis of CAG instability and its role in the disease process.
Return to "Research" section or table of contents
Fetal Tissue Research
A relatively new field in biomedical research involves the use of brain tissue grafts to study, and potentially treat, neurodegenerative disorders. In this technique, tissue that has degenerated is replaced with implants of fresh, fetal tissue, taken at the very early stages of development. Investigators are interested in applying brain tissue implants to HD research. Extensive animal studies will be required to learn if this technique could be of value in individuals with HD.
Return to "Research" section or table of contents
Clinical Studies
Scientists are pursuing clinical studies that may one day lead to the development of new drugs or other treatments to halt the disease's progression. Examples of NINDS-supported investigations, using both asymptomatic and symptomatic individuals, include:
Genetic studies on age of onset, inheritance patterns, and markers found within families.These studies may shed additional light on how HD is passed from generation to generation.
Studies of cognition, intelligence, and movement. Studies of abnormal eye movements, both horizontal and vertical, and tests of patients' skills in a number of learning, memory, neuropsychological, and motor tasks may serve to identify when the various symptoms of HD appear and to characterize their range and severity.
Clinical trials of drugs. Testing of various drugs may lead to new treatments and at the same time improve our understanding of the disease process in HD. Classes of drugs being tested include those that control symptoms, slow the rate of progression of HD, and block effects of excitotoxins, and those that might correct or replace other metabolic defects contributing to the development and progression of HD.
Return to "Research" section or table of contents
Imaging
NINDS-supported scientists are using positron emission tomography (PET) to learn how the gene affects the chemical systems of the body. PET visualizes metabolic or chemical abnormalities in the body, and investigators hope to ascertain if PET scans can reveal any abnormalities that signal HD. Investigators conducting HD research are also using PET to characterize neurons that have died and chemicals that are depleted in parts of the brain affected by HD.
Like PET, a form of magnetic resonance imaging (MRI) called functional MRI can measure increases or decreases in certain brain chemicals thought to play a key role in HD. Functional MRI studies are also helping investigators understand how HD kills neurons in different regions of the brain.
Imaging technologies allow investigators to view changes in the volume and structures of the brain and to pinpoint when these changes occur in HD. Scientists know that in brains affected by HD, the basal ganglia, cortex, and ventricles all show atrophy or other alterations.
Return to "Research" section or table of contents
How Can I Help?
In order to conduct HD research, investigators require samples of tissue or blood from families with HD. Access to individuals with HD and their families may be difficult however, because families with HD are often scattered across the country or around the world. A research project may need individuals of a particular age or gender or from a certain geographic area. Some scientists need only statistical data while others may require a sample of blood, urine, or skin from family members. All of these factors complicate the task of finding volunteers. The following NINDS-supported efforts bring together families with HD, voluntary health agencies, and scientists in an effort to advance science and speed a cure.
Return to table of contents
The NINDS-sponsored HD Research Roster at the Indiana University Medical Center in Indianapolis, which was discussed earlier, makes research possible by matching scientists with patient and family volunteers. The first DNA bank was established through the roster. Although the gene has already been located, DNA from individuals who have HD is still of great interest to investigators. Of continuing interest are twins, unaffected individuals who have affected offspring, and individuals with two defective HD genes, one from each parenta very rare occurrence. Participation in the roster and in specific research projects is voluntary and confidential. For more information about the roster and DNA bank, contact:
Indiana University Medical Center
Department of Medical and Molecular Genetics
Medical Research and Library Building
975 W. Walnut Street
Indianapolis, IN 46202-5251
(317) 274-5744 (call collect)
Return to table of contents
Brain tissue is also critical to the HD research effort, and many individuals are willing to donate their brains and other organs to research after they die. The NINDS supports two national human brain specimen banks, one at the Wadsworth Veterans Administration Medical Center in Los Angeles, and the other at McLean Hospital near Boston. These banks supply investigators around the world with tissue not only from individuals with HD but also from those with other neurological or psychiatric diseases. Both banks need brain tissue to enable scientists to study these disorders more intensely. Prospective donors should contact:
Wallace W. Tourtellotte, M.D., Ph.D.
Director, National Neurological Research Specimen Bank
VA Wadsworth Medical Center Neurology Research
Wilshire & Sawtelle Boulevards
Los Angeles, CA 90073
(310) 268-3536 (call collect)
Francine M. Benes, M.D., Ph.D.
Director, Harvard Brain Tissue Resource Center
Mailman Research Center, McLean Hospital
115 Mill Street
Belmont, MA 02178
(617) 855-2400 (call collect)
(800) 272-4622
Return to table of contents
What is the Role of Voluntary Organizations?
Private organizations have been a mainstay of support and guidance for at-risk individuals, people with HD, and their families. These organizations vary in size and emphasis, but all are concerned with helping individuals and their families, educating lay and professional audiences about HD, and promoting medical research on the disorder. Some voluntary health agencies support scientific workshops and research and some have newsletters and local chapters throughout the country. These agencies enable families, health professionals, and investigators to exchange information, learn of available services and benefits, and work toward common goals. The organizations listed in the Information Resources section of this brochure welcome inquiries from the public.
Return to table of contents
Glossary
Return to table of contents
Current Medical Treatment of Huntington's Disease
(Abstract: Huntington's Disease Workshop, Houston, TX)
Joseph Jankovic, M.D.The first step in the management of patients with Huntington s disease (HD) is education of the patient and the family about the nature of the disease and the prognosis. This must be coupled with skilled genetic and psychological counseling. In the Baylor College of Medicine Huntington s Disease Clinic, we also provide educational material and referral resources. Because of the complex and heterogeneous nature of the disorder, medical therapy must be individualized and tailored to specific needs of the patient. The medications are targeted to control the most troublesome symptoms.
Depression, commonly seen even in early stages of the disease, is partly biological and partly situational arising from the realization of impending progressive functional impairment. Supportive and understanding attitude from family and friends, while extremely important, may not be sufficient and most patients will eventually require medical therapy. Tricyclic antidepressants, such as amitriptyline, imipramine and nortriptyline, and serotoninergic agents, such as fluoxetine and sertraline, are used most commonly. The tricyclics, when given at night, have the advantage of helping insomnia and by stimulating appetite they may prevent weight loss, frequently seen in patients with HD. The serotonergic drugs are helpful in patients who, in addition to depression, exhibit obsessive compulsive disorder. Anxiolytics, such as diazepam, alpralozam, and clonazepam, may be helpful to control agitation. We also sometimes use carbamazepine, valproate, and lithium to help control manic behavior. Impulse control problems may respond to a trial with clonidine or propranolol. Rarely, electroconvulsive therapy is sometimes required in patients with medically intractable depression.
Psychosis may improve with dopamine receptor blocking drugs (neuroleptics), such as haloperidol, pimozide, fluphenazine and thioridazine, but these drugs can induce tardive dyskinesia and adverse effects and should be used only if absolutely needed to control symptoms. Clozapine, an atypical antipsychotic drug that does not cause tardive dyskinesia, may be a useful alternative to the typical neuroleptics, but its high cost, risk of agranulocytosis, and other potential side effects limit its use.
Neuroleptics are the most effective drugs in the treatment of chorea. The dopamine blocking drugs, however, may cause tardive dyskinesia. Monoamine depleting drugs, such as reserpine and tetrabenazine, have the advantage that they do not cause tardive dyskinesia. In our experience, tetrabenazine is the most effective suppressant of chorea, but this drug is categorized as investigational and as such is not readily available in the U.S.. Both classes of neuroleptics may cause or exacerbate depression, sedation, akathisia and parkinsonism.
It has been suggested, that in HD, as a result of prolonged excitatory neurotransmission, certain neurons become "exhausted" and switch from aerobic to anaerobic, glycolytic metabolism, leading to the production and accumulation of lactate. Recent studies have demonstrated that administration of agents which improve energy metabolism, such as coenzyme Q10 and nicotinamide, may protect animals against toxicity produced by malonate, a complex II inhibitor. Although coenzyme Q10 and nicotinamide may eventually prove to exert neuroprotective effect in humans, only symptomatic treatment is currently available for HD patients. Whether blocking glutamate release from the presynaptic terminals by drugs such as lamotrigine or whether other antiglutamatergic drugs will be effective in slowing down the otherwise inexorable progression of the disease awaits further studies, some of which are already in progress. Baclofen, a putative GABA agonist, provides neither symptomatic nor protective effects in HD.
Implantation of nerve-growth factor-producing fibroblasts into the rat striatum appears to protect these animals against neurotoxic effects of excitatory amino acids quinolate and quisqualate. Whether intrastriatal implantations of genetically engineered or fetal cells will be useful in the treatment of HD awaits the results of further animal and pre-clinical studies.
Clinical Genetics of Huntington's Disease(Abstract: Huntington's Disease Workshop,
Houston, TX
Updated by Tetsuo Ashizawa, M.D. 3/20/97) Stuart K. Shapira, M.D., Ph.D.Huntington's disease was long
known to be a genetic condition, in that it could run in families and appeared to be passed
directly from parent to child, without skipping generations or showing a preference for
affecting one sex more than the other. The pattern of inheritance in which the disease runs
in families is known as autosomal dominant inheritance. All genetic information, excepting
that which is included in the sex chromosomes of a male, occurs in two copies within cells,
and autosomal dominant disorders occur when there is an alteration (mutation) in one of
the two copies of a particular segment of genetic information (gene). In other words, a
mutation in one of the two genes is necessary and sufficient to cause the disorder in an
individual. With regard to the mutant gene, there is a 50% chance of it being passed to a
given offspring, and the two sexes are equally likely to inherit the mutant gene and be
affected with the disorder. If a child inherits the copy of the gene pair that does not have
the mutation (also a 50% chance), then the child should not develop the disorder, and will
also not pass on the condition to his or her children.
All individuals who inherit a mutant Huntington's disease gene will develop signs and symptoms of the condition, if they live long enough. The disorder does not skip generations, but the age at which symptoms of the disorder develop can vary widely among individuals, even within the same family. However, it was frequently noted that if a man has Huntington's disease, his affected children can show symptoms of the condition at much younger ages than the symptoms first developed in him. In fact his children might even develop their first symptoms of the disease before 10 years of age. This phenomenon of a genetic condition running in families whereby a mildly affected parent could have a more severely affected child is called "anticipation". Specifically, for Huntington's disease, it was observed that as the condition was passed from parent to child, and more specifically from an affected father to a child, the symptoms of the condition could become apparent at younger and younger ages in each successive generation within the family. The anticipation phenomenon was nicely explained when the Huntington's disease gene was found and the mutation within the gene that caused the disease was identified.
In 1983, the defective genetic information (gene) responsible for Huntington's disease was found to be located on chromosome #4, and in 1986, the gene was localized to the end of the short arm (top) of the chromosome. In 1993, the Huntington's disease gene was finally found, and the source of the mutation (alteration) in the gene that leads to the disease was identified. When the composition of the genetic information within the Huntington's disease gene was determined, it was found that near the beginning of the genetic information comprising the gene was a series of repeated units of information, known as CAG repeats; CAG represents a three-chemical unit of the genetic information (trinucleotide) that is repeated a specified number of times in a tandem array within the gene. When the Huntington's disease genes were examined from individuals who do not have the disorder, it was found that these genes had less than 29 copies of the CAG trinucleotide repeat within each gene. However, individuals that developed Huntington's disease would have one normal gene with a normal number of the CAG repeats (less than 29 copies), and their other gene (the mutant gene) would have an expanded number of CAG repeats (generally greater than 35, with most mutant genes having between 40 and 55 CAG repeats). Rarely will the CAG repeat length be more than 70, but it can be over 100. Some individuals with 36 to 39 CAG repeats do not develop Huntington's disease even at very old age (greater than 80 years old). Thus, this range is considered as the "reduced penetrance range". Individuals with 29 to 35 CAG repeats have not been documented with Huntington's disease. However, some of these individuals may produce children with the disease. CAG repeats in this range have shown instability of the repeat size which may result in expansions to greater than 40 CAG repeats in affected children. Therefore, 29 to 35 CAG repeats are considered "mutable normal" range.
It was subsequently shown that there was a significant inverse correlation between the age at which an individual's symptoms of Huntington's disease first become apparent, and the number of CAG repeats in the mutant gene. In other words, the more CAG repeats, often the earlier the age that symptoms first appear. Individuals with childhood (<10 years of age) and juvenile (11-20 years of age) onset of the symptoms of Huntington's disease were found to have the larger CAG trinucleotide repeat lengths in their mutant Huntington's disease gene. However, the CAG repeat size is not a clinically useful predictor of either the age of onset or the rate of progression in individual patients.
It was also observed that children and juveniles with Huntington's disease had a much higher risk (75-80% risk) of having inherited their mutant gene with the expanded number of CAG repeats from their father. This observation helped to explain the anticipation phenomenon in this condition. When the gene is passed through the germ cells (eggs or sperm) to the next generation, the number of CAG repeats may increase, hence producing a more severely affected offspring from a less severely affected parent. For Huntington's disease, this was particularly apparent when the mutant gene was passed via the father to the child, indicating a sex-specific effect on the likelihood of expansion of the CAG repeat when passed from an affected father to a child. Though expansions of the CAG repeat do occur when passed from an affected mother to a child, generally the repeat does not expand to the number of CAG repeats that causes juvenile Huntington's disease unless the mother of the child has juvenile onset of the condition, herself.
With the identification if the Huntington's disease gene and the characterization of the
CAG trinucleotide repeat expansion that leads to the disorder, there has been a major
advance in predictive testing for the condition. Direct testing for the CAG repeat length can
not only identify individuals who will develop the disorder, it can identify those individuals
who have not inherited the mutant gene from a parent. In addition, direct mutation analysis
of the CAG repeat length can be used for prenatal testing for Huntington's disease, if
parents choose this as an option.
Clinical Presentations of Huntington's Disease (Abstract: Huntington's Disease Workshop, Houston, TX) Joseph Jankovic, M.D.George Huntington published "On Chorea" in 1872 and the eponym "Huntington's Chorea" was soon adapted in the literature to draw attention to chorea (derived from Latin 'choreus' = dance and Greek 'choros' = chorus) as the clinical hallmark of this neurodegenerative disorder. However, since there are many other manifestations of the disease and chorea may not even be present, the term "Huntington's Disease" (HD) seems more appropriate for this heredodegenerative disorder. Chorea consists of involuntary, continuous, abrupt, rapid, brief, unsustained, irregular movements that flow randomly from one body part to another. Patients can partially and temporarily suppress the chorea and frequently "camouflage" some of the movements by incorporating them into semipurposeful activities (parakinesia). The inability to maintain voluntary contraction (motor impersistance), such as tongue protrusion or manual grip (milk-maid grip), is a characteristic feature of chorea and results in dropping of objects and clumsiness. Chorea should be differentiated from pseudochoreoathetosis , a movement disorder phenomenologically similar to chorea or athetosis (slow chorea) due to loss of position sense (proprioception). Muscle stretch reflexes are often "hung-up" and "pendular". Affected patients typically have a peculiar, irregular, and dance-like gait. Other motor symptoms include dysarthria, dysphagia, postural instability, ataxia, dystonia, myoclonus, and tics. In a study of 593 members of a large kindred in Venezuela, the generation of fine motor movements and of rapid eye saccades was found impaired in about 50% of at-risk individuals. Since at-risk individuals with these findings were more likely to develop overt HD within several years, these abnormalities were thought to represent the earliest clinical manifestations of the disease. Besides chorea, the other two components of the HD triad include cognitive decline and various psychiatric symptoms. The neurobehavioral symptoms typically consist of personality changes, apathy, social withdrawal, agitation, impulsiveness, depression, mania, paranoia, delusions, hostility, hallucinations or psychosis. Cognitive changes, manifested chiefly by loss of recent memory, poor judgement, impaired concentration and acquisition, occur in nearly all patients with HD; but some patients with late-onset chorea never develop dementia. Tasks requiring psychomotor or visuospatial processing, such as skills required by Trail Making B and Stroop Interference Test, are impaired early in the course of the disease and deteriorate at a more rapid rate than memory impairment. Although neurobehavioral symptoms may precede motor disturbances, neuropsychological tests do not differentiate between presymptomatic persons who are positive for the HD marker from those who are negative. About 10% of HD cases have their onset before age 20, but the typical peak age at onset is in the 4th and 5th decade. Young-onset patients usually inherit the disease from their father while older- onset patients are more likely to inherit the gene from their mother. Juvenile HD (onset of symptoms before 20 years) typically presents with the combination of progressive parkinsonism, dementia, ataxia, and seizures. In contrast, adult HD usually presents with the insidious onset of clumsiness and adventitious movements which may be wrongly attributed to simple nervousness. Slowness of movement (bradykinesia) is usually evident in patients with the rigid form of HD, but when it coexists with chorea it may not be fully appreciated on a routine examination. While bradykinesia is most pronounced in the rigid-akinetic patients, it is also evident in patients with the typical choreic variety of HD. When bradykinesia predominates, the patients exhibit parkinsonian findings some of which may be subtle. Micrographia may be one manifestation of underlying parkinsonism; when chorea predominates the handwriting is characterized by macrographia. Bradykinesia in HD may be an expression of "post-synaptic parkinsonism" and possibly explains why a reduction in chorea with anti-dopaminergic drugs rarely improves overall motor functioning and indeed may cause an exacerbation of the motor impairment. The natural course and prognosis of HD is quite variable: duration of illness from onset to death is about 15 years for adult HD and 8 to 10 years for the juvenile variant. Clinical-pathological studies have demonstrated strong inverse correlation between the age at onset and the severity of striatal degeneration. A review of clinical and pathological data in HD patients showed that patients with juvenile-adolescent onset had much more aggressive progression of the disease than patients with onset in middle and late life. Progressive motor dysfunction, dementia, dysphagia, and incontinence eventually lead to institutionalization and death from aspiration, infection, and poor nutrition.
Advances in Understanding of the Disease Mechanisms(Abstract: Huntington's Disease Workshop, Houston, TX)
Tetsuo Ashizawa, M.D.In Huntington's disease (HD), uncontrollable involuntary movements, psychiatric abnormalities and a loss of intellectual functions (dementia) are the three major manifestations. Involuntary movements, such as chorea, result from abnormalities in the structures called basal ganglia which are located deep in the brain and regulate motor movements. One of these structures called striatum shows a decreased volume in HD. The atrophy is due to degeneration of a particular subpopulation of the neurons (brain cells with electrical activities) called medium-size spiny neurons located within the striatum. Dementia and psychiatric abnormalities are due to degeneration of neurons outside the basal ganglia. A loss of neurons in the cerebral cortex (the surface layers of the brain) is particularly prominent in HD.
The mechanism of the degeneration is not fully understood. However, the final process of brain cell death appears to be mediated by a class of amino acids (called excitatory amino acids) released from other neurons in which excessive excitation of neurons causes "exhaustion" of the neurons and eventually leads to cell death, especially when the neurons already suffer from a disease process. This phenomenon is called "excitotoxic cell death." Injecting chemical compounds that activate the excitotoxic receptors into animals produces a selective loss of neurons closely resembling HD. Biochemical changes and behavioral changes in these animals are also very similar to HD. These studies suggest that excitotoxic cell death may play an important role in HD.
The energy metabolism appears to be affected in the medium-size spiny neurons in the striatum in HD patients. The abnormal energy metabolism quickly "exhausts" the neurons, making them susceptible to excitotoxicity. Mitochondria are the power house of cells and produce energy by a chain of chemical reactions called oxidative phosphorylation. Inhibitors of oxidative phosphorylation such as 3-nitropropionic acid (3NP) cause a disease with similar clinical signs and increased lactate levels similar to those seen in HD. Interestingly, 3NP causes damage specifically in the medium size spiny neurons and, consequently, in the cortical neurons. Thus, abnormal energy metabolism may contribute to the disease-causing mechanisms in HD.
The key question is how the genetic abnormality in HD can lead to these changes in the brain? Since CAG codes for glutamine, the protein coded by the HD gene, called huntingtin, should have a tract of tandemly repeated glutamines. In HD, the expanded CAG repeat tract should be translated into an expanded glutamine repeat tract. In fact, enlarged huntingtin protein molecules were found in HD patients and they appeared to contain expanded glutamine repeats. However, in addition to this mutant gene, HD patients have the other chromosome 4 which has normal size CAG repeat. Therefore, they have the huntingtin protein with a normal glutamine tract in an amount of 50% of normal individuals. For most proteins, cells can function normally if there is 50% of the normal amount, and this is true for huntingtin. Patients in whom a piece of DNA containing the entire HD gene is deleted from one of the chromosomes 4 do not have any signs of HD. Transgenic mice (genetically engineered mice) in which one of the two HD genes has been "knocked out" also fail to show signs of HD. Thus, an idea that the abnormal huntingtin with an expanded tract of glutamine repeat gains new function was introduced to explain these observations. If this gain-of-function theory is right, what is the new function? The amino acid sequence of huntingtin has no resemblance to any known protein. As a result, what normal huntingtin does in cells is unknown. Furthermore, this gain of function must significantly affect only certain neurons in the brain, since no other tissues are abnormal in HD despite the widespread presence of huntingtin protein among various tissues.
Experimental data have suggested that huntingtin does interact with other cellular proteins, including huntingtin associated protein 1 (HAP-1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), apopain and ubiquitin conjugating enzyme (hE2-25K). In the presence of calcium, huntingtin also interacts indirectly with calmodulin. Among all these interactions, HAP-1, and apopain show huntingtin-specific interactions, while GAPDH interacts with other molecules containing a polyglutamine tract. The interactions of HAP-1, GAPDH, apopain and calmodulin with huntingtin are stronger when the polyglutamine tract is longer, whereas the huntingtin-E2-25K interaction is not obvious to be dependent on the CAG length. Since all these proteins are expressed not only in the striatum but also in other parts of the brain, regional distributions of these proteins are an unlikely cause of the selective neuronal death in HD. However, by postulating another tissue-specific variable, we may be able to explain why the medium-sized spiny neurons are more vulnerable. Interesting hypotheses have been proposed for each huntingtin-interacting protein. Preliminary reports suggested that there will be other huntingtin-interacting proteins in addition to those described above. However, whether any of these proteins actually play a significant role in the disease mechanisms of HD remains to be further investigated. An alternative pathophysiological mechanism involving proteins that bind to CAG repeats within the huntingtin RNA have been postulated.Transgenic mice, which have multiple and random integrations of a transgene containing ~160 CAG repeat, exhibited clinical and pathological features similar to HD. It is unknown whether these animal models truly reflect the disease mechanisms of HD. However, such animal models may provide important insights into understanding the disease mechanisms in HD.
Last Updated on December 5, 1998 by Dr.danil hammoudi

{kind=link}