

USMLE STEP 2 PART VIII
CLINICAL DISORDERS OF LIPOPROTEIN METABOLISM:
- HYPERLIPIDEMIAS
- HYPOLIPIDEMIAS:
-
-
- HYPOBETALIPOPROTEINEMIA =BASSEN - KORNZWEIG SYNDROME
- ALPHA-LIPOPROTEIN DEFICIENCY = TANGIER DISEASE
-
|
HYPERLIPIDEMIAS |
HYPOLIPIDEMIAS |
| can be primary [ classification ==> electrophoretic patterns
after 12h fast]
can be secondary to underlying disease [THYROID-LIVER-KIDNEY] |
1/HYPO-BETA LIPOPROTEINEMIA=BASSEN KORNZWEIG SYNDROME
Rare genetic disorder recessive inheritance characterized by neurologic symptoms:
|
MAIN LIPID COMPONENTS, SITE OF SYNTHESIS AND FUNCTION OF LIPOPROTEINS:
| LIPOPROTEINS | LIPIDS | SITE OF SYNTHESIS | FUNCTION |
| CHYLOMICRONS | TRIGLYCERIDES | INTESTINE | TRANSPORTATION OF DIETARY [EXOGENOUS] TRIGLYCERIDES TO THE LIVER |
| VLDL | TRIGLYCERIDES | LIVER
INTESTINE |
TRANSPOTATION OF ENDOGENOUS TRIGLYCERIDES |
| LDL | CHOLESTEROL | INTRAVASCULAR
FINAL PRODUCT OF VLDL BREAKDOWN |
TRANSPORTATION OF CHOLESTEROL TO THE PERIPHERAL CELLS |
| HDL | CHOLESTEROL
PHOSPHOLIPIDS |
LIVER
INTESTINE INTRAVASCULAR FINAL PRODUCTION OF BREAKDOWN OF CHYLOMICRONS AND VLDL |
TRANSPORTATION OF CHOLESTEROL FROM PERIPHERAL CELLS BACK TO THE LIVER [ELIMINATION AS BILE ACIDS VIA INTESTINE] |
CLASSIFICATION OF PRIMARY HYPERLIPIDEMIAS
| TYPE | GENETIC CLASSIFICATION | Genetic form | Elevated plasma lipid | Elevated plasma lipoprotein | Risk of atherosclerosis | trt |
| I | FAMILIAL LIPOPROTEIN LIPASE DEFICIENCY | AR | TRIAGLYCEROLS | CHYLOMICRONS | Not clearly increased | LOW FAT DIET |
| IIA | FAMILIAL HYPERCHOLESTEROLEMIA | AD | CHOLESTEROL | LDLs | Very high especially in coronary artery | LOW CHOLESTEROL DIET
CHOLESTYRAMINE POSSIBLE SURGERY |
| IIB | TRIACYLGLYCEROL CHOLESTEROL | LDLs AND VLDLs | ||||
| III | FAMILIAL DYS-BETA LIPOPROTEINEMIA | UNCERTAIN | TRIACYLGLYCEROL CHOLESTEROL | BETA-VLDL | VERY HIGH [PERIPHERAL VESSELS] | LOW CHOLESTEROL
LOW CALORIES DIET CLOFIBRATE |
| IV | FAMILIAL HYPERTRIGLYCERIDEMIA | HETEROGENOUS COMMON | TRIACYLGLYCEROL | VLDL | POSSIBLE | LOW FAT
LOW CALORIE DIET NO ALCOHOL NIACIN |
| V | HETEROGENOUS | TRIACYGLYCEROL | CHYLOMICRONS
VLDLs |
NOT CLEARLY INCREASED | LOW FAT
LOW CALORIE DIET NIACIN |
AR: AUTOSOMAL RECESSIVE
AD: AUTOSOMAL DOMINANT
GLYCOGEN STORAGE DISEASE
| MC ARDLE DISEASE |
|
| CORI DISEASE: |
|
|
CLASSIFICATION BY SYMPTOMS VON GIERKE DISEASE: |
1/ HEPATIC -HYPOGLYCEMIC EFFECTS
2/MUSCLE PROBLEMS:
-PAIN - CRAMP- MYOGLOBINURIA
|


The Allied Diseases Profiled
TAY-SACHS AND THE ALLIED DISEASES ARE GENETIC CONDITIONS CLASSIFIED as storage diseases. They are caused by the abnormal accumulation, or storage, of certain waste products in the cells or tissues of affected individuals. As these products build up, cells become damaged and gradually lose their ability to function properly, causing disease symptoms. While the specific clinical courses of these related disorders differ, there are certain commonalities, and children and adults affected with Tay-Sachs or any of the allied diseases share many issues associated with chronic, progressive illness.
The chart below provides a quick reference for the major characteristics of the allied diseases. Underlined words are links to more information on this site or elsewhere on the Internet. The Omim # refers to the catalogue citation on the Online Mendelian Inheritance In Man, the hypertext version of Victor McCusick's landmark catalogue of human genetic disease.
Additionally, the following Allied Diseases are profiled in more depth in their own sections:
- Tay-Sachs Disease
- Late-Onset Tay-Sachs Disease
- Sandhoff Disease
- Fabry Disease
- Gaucher Disease
- Niemann-Pick Disease
- Canavan Disease
This information is provided in response to a growing demand for knowledge and in the hopes of increasing awareness and understanding of these rare, but often devastating, diseases.
Tay-Sachs & the Allied Diseases
Category Index:
Disorders of
lipid and sphingloid degradation
Disorders of
mucopolysaccharide degradation
Disorders
of glycoprotein degradation
Other
lysosomal storage disorders
Non-lysosomal
diseases
| Disease | Enzyme Defect | Prenatal Diagnosis? | Carrier Testing? | Chromosome Location | Inheritance Pattern | |
| A. Lysosomal Storage Disorders | ||||||
| 1) Disorders of lipid and sphingloid degradation | ||||||
| GM1 Gangliodsidosis | b-Galactosidase | Yes | Yes | 3 | AR | |
| Tay-Sachs Disease | b-Hexosaminidase A | Yes | Yes | 15 | AR | |
| Sandhoff Disease | b-Hexosamindase A&B | Yes | Yes | 5 | AR | |
| GM2 Gangliosidosis: AB Variant | GM2 Activator Protein | Yes | No | 5 | AR | |
| Fabry Disease | a-Galactosidase A | Yes | Yes | X | X-Linked | |
| Gaucher Disease | Glucocerebrosidase | Yes | Yes | 1 | AR | |
| Metachromatic Leukodystrophy | Arylsulfatase A | Yes | Yes | 22 | AR | |
| Krabbe Disease | Galactosylceramidase | Yes | Yes | 14 | AR | |
| Niemann-Pick, Types A and B | Acid Sphingomyelinase | Yes | Yes | 18 | AR | |
| Niemann-Pick, Type C | Cholesterol Esterification Defect | Yes | Yes | 18 | AR | |
| Farber Disease | Acid Ceramidase | Yes | Yes | ? | (AR) | |
| Wolman Disease | Acid Lipase | Yes | Yes | 10 | AR | |
| Cholesterol Storage Disease | Acid Lipase | Yes | Yes | 10 | AR | |
| Back to top | ||||||
| 2) Disorders of mucopolysaccharide degradation | ||||||
| Hurler Syndrome (MPS III) |
a-L-Iduronidase |
Yes |
Yes |
4 |
AR | |
| Scheie Syndrome (MPS IS) |
a-L-Iduronidase | Yes | Yes | 4 | AR | |
| Hurler-Scheie (MPS IH/S) |
a-L-Iduronidase | Yes | Yes | 4 | AR | |
| Hunter Syndrome (MPS II) |
Iduronate Sulfatase | Yes | Yes | X | X-Linked | |
| Sanfilippo A (MPS IIIA) |
Heparan N-Sulfatase | Yes | ? | 17 | AR | |
| Sanfilippo B (MPS IIIB) |
a-N-Acetylglucosaminidase | Yes | Yes | 17 | AR | |
| Sanfilippo C (MPS IIIC) |
Acetyl-CoA-Glucosaminide Acetyltransferase | Yes | ? | 14 | AR | |
| Sanfilippo D (MPS IIID) |
N-Acetylglucosamine -6-Sulfatase | Yes | ? | 12 | AR | |
| Morquio A (MPS IVA) |
Galactosamine-6-Sulfatase | Yes | Yes | 16 | AR | |
| Morquio B (MPS IVB) |
b-Galactosidase | Yes | Yes | 3 | AR | |
| Maroteaux-Lamy (MPS VI) |
Arylsulfatase B | Yes | Yes | 5 | AR | |
| Sly Syndrome (MPS VII) |
b-Glucuronidase | Yes | Yes | 7 | AR | |
| Back to top | ||||||
| 3) Disorders of glycoprotein degradation | ||||||
| Mannosidosis | a-Mannosidase | Yes | ? | 19 | AR | |
| Fucosidosis | a-L-Fucosidase | Yes | ? | 1 | AR | |
| Asparylglucosaminuria | N-Aspartyl- b-Glucosaminidase | Yes | Yes | 4 | AR | |
| Sialidosis (Mucolipidosis I) | a-Neuraminidase | Yes | ? | 20 | AR | |
| Galactosialidosis | Lysosomal Protective Protein | Yes | ? | 20 | AR | |
| Schindler Disease | a-N-Acetyl- Galactosaminidase | Yes | Yes | 22 | AR | |
| Back to top | ||||||
| 4) Other lysosomal storage disorders | ||||||
| Batten Disease (Juvenile Neuronal Ceroid Lipofuscinosis) | Unknown | Yes | ? | 16 | AR | |
| Infantile Neuronal Ceroid Lipofuscinosis) | Palmitoyl-Protein Thioesterase | Yes | Yes | 4 | AR | |
| Pompe Disease | Acid-a1, 4-Glucosidase | Yes | Yes | 17 | AR | |
| Mucolipidosis II (I-Cell Disease) | N-Acetylglucosamine-1- Phosphotransferase | Yes | Yes | 4 | AR | |
| Mucolipidosis III (Pseudo-Hurler Polydystrophy) | Same as ML II | Yes | Yes | 4 | AR | |
| Mucolipdosis IV | Unknown | ? | No | ? | (AR) | |
| Cystinosis | Cystine Transport Protein | Yes | Yes | 17 | AR | |
| Salla Disease | Sialic Acid Transport Protein | Yes | ? | 6 | AR | |
| Infantile Sialic Acid Storage Disease | Sialic Acid Transport Protein | Yes | ? | 6 | AR | |
| Saposin Deficiencies | Saposins A, B, C or D | Yes | No | 10 | AR | |
| Back to top | ||||||
| B. Non-Lysosomal Diseases | ||||||
| Abetalipoproteinemia | Microsomal Triglyceride Transfer Protein | Yes | ? | 4 | AR | |
| X-Linked Adrenoleuk- odystrophy | Peroxisomal Membrane Transfer Protein | Yes | Yes | X | X-Linked | |
| Refsum Disease | Phytanic Acid a-Hydroxylase | Yes | ? | ? | (AR) | |
| Canavan Disease | Aspartoacylase | Yes | Yes | 17 | AR | |
| Cerebrotendinous Xanthromatosis | Sterol-27-Hydroxlase | No | Yes | 2 | AR | |
| Pelizaeus Merzbacher Disease | Lipophlin | Yes | Yes | X | X-Linked | |
| Tangier Disease | Apo-Gln-1 | No | No | ? | (AR) | |
| Back to top | ||||||
GENETICS OF G6PD DEFICIENCY
t is important to learn about the genetics of
G6PD deficiency since this determines whether someone will be affected by this
condition. In humans, there are 23 pairs of chromosomes which direct various
physical and metabolic traits. One of the 23 pairs of chromosomes is the X and
Y- chromosome pair (also known as the sex chromosomes) which determine what sex
an individual will be, among other things. The X-chromosome is especially
important because it carries genes that are critical to human survival. An
important gene located on the X-chromosome is the gene for the G6PD enzyme
(Scriver et al., 1995). The general location of the G6PD gene on the
X-chromosome is shown in figure 1
; it is located at the q28 locus (Pai et al., 1980).
Any gene located on the X-chromosome is called an X-linked gene (Pai et al., 1980). All X-linked genetic conditions, such as G6PD deficiency, are more likely to affect males than females. G6PD deficiency will only manifest itself in females when there are two defective copies of the gene in the genome. As long as there is one good copy of the G6PD gene in a female, a normal enzyme will be produced and this normal enzyme can then take over the function that the defective enzyme lacks. When a certain heritable trait is expressed in such a manner, it is a called a recessive trait. In males, however, where there is only one X-chromosome, one defective G6PD gene is sufficient to cause G6PD deficiency.
G6PD deficiency is known to have over 400 variant alleles, or different forms of the same gene (Beutler et al., 1990). Ernest Beutler, one of the leading researchers on G6PD deficiency, provides an up-to-date list of all known G6PD variants in the journal which he edits: Blood, Cells, Molecules, and Diseases(BCMD). A mutant G6PD enzyme may be different from person to person; mutations can be in the form of point mutations or can range from one to several base pair deletions as well as replacements in the DNA (Scriver et al., 1995). Different populations have different types of mutations, but within a specific population, common mutations are usually shared. For example, in Egypt there exists only one type of allele, called the "Mediterranean" variant, among the population, whereas in Japan there is a different variant with a different type of mutation prevalent within that population, this one called the "Japan" variant (Scriver et al., 1995).
With regards to the demographics of G6PD deficiency, figure 2 shows that most of the affected individuals reside in Africa, the Middle East, and Southeast Asia. African Americans and some isolated tribes in Africa and Southeast Asia exhibit the highest frequency of incidence for any given population; a defective enzyme can be found in as many as one in four people among these populations (Scriver et al., 1995).
 CLINICAL ASPECTS OF G6PD DEFICIENCY
CLINICAL ASPECTS OF G6PD DEFICIENCY
hen the red blood cell can no longer transport
oxygen effectively throughout the body, a condition called hemolytic anemia
arises. In addition to hemolytic anemia, G6PD deficient individuals can expect
several other clinical manifestations of their condition. These include neonatal
jaundice, abdominal and/or back pain, dizziness, headache, dyspnea (irregular
breathing), and palpitations (Cecil, 1992). Only neonatal jaundice and hemolytic
anemia will be discussed here, since these are the two major pathologies
associated with G6PD deficiency (see Cecil, 1992, and Scriver et al., 1995, for
a discussion of the other clinical manifestations of G6PD deficiency).
NEONATAL JAUNDICE
One of the problems experienced by G6PD
deficient individuals presents itself immediately after birth. Neonatal jaundice
is a common condition in all newborns, but when it persists, G6PD deficiency is
suspected. Neonatal jaundice is a yellowish discoloration of the whites of the
eyes, skin, and mucous membranes caused by deposition of bile salts in these
tissues. This is a direct result of insufficient activity of the G6PD enzyme in
the liver. In some cases, the neonatal jaundice is severe enough to cause death
or permanent neurologic damage (Beutler, 1994).
HEMOLYTIC ANEMIA
Hemolytic anemia is another condition which may
cause problems for G6PD deficient individuals. An anemic response can be induced
in affected individuals by certain oxidative drugs, fava beans, or infections
(Beutler, 1994). Death ensues if the hemolytic episode is not properly treated.
In order to prevent a severe reaction or even death, G6PD deficient individuals
are prohibited from taking certain drugs; a list of drugs that are commonly
reported in the literature as inducing hemolysis in G6PD deficient individuals
appears in Table 2. The common
theme shared among all of these drugs is that they are oxidizing agents.In G6PD
deficient individuals, oxidative stress may result in the denaturation, or
unfolding, of the hemoglobin molecule, the principal oxygen carrying molecule
inside the red blood cell. This results in the loss of biological function with
respect to hemoglobin and leads to the inability of the red blood cell to
effectively transport oxygen throughout the body (Yoshida & Beutler, 1986).
For reasons still unknown, some G6PD deficient individuals do not exhibit
drug-induced hemolytic anemia when exposed to certain drugs on this list; of
course, a physician should always be consulted before any medications are taken.
Primaquine, one of the first anti-malarial drugs, was the first drug to be implicated in inducing an anemic response (Carson et al., 1956). All known anti-malarial drugs are contra-indicated for G6PD deficient individuals (see Table 2); however, in cases of acute uncomplicated malaria, most anti-malarial drugs can be safely administered (Baird, personal correspondence). It is interesting to note that a deficiency in G6PD has been shown to sometimes confer a resistance to the malaria-causing parasite, Plasmodium falciparum (Scriver et al., 1995). This resistance is due to the fact that the parasite selectively infects red blood cells. In G6PD deficient red blood cells, an essential metabolite for the survival of the parasite is present in insufficient quantities. This is due to decreased activity of G6PD within these cells which ultimately leads to the death of the parasite (Farid, personal interview).
In addition to drug-induced hemolytic anemia, there is also fava bean- induced hemolytic anemia--called favism. Fava beans were the first, and only food product, to be implicated in inducing an anemic response in G6PD deficient individuals (Scriver et al., 1995). Inhaling the pollen of the fava bean plant can also induce hemolysis in favic individuals. Since some G6PD deficient individuals are allergic to fava beans, the deficiency is therefore sometimes referred to as favism (THE FAVISM HOMEPAGE). Favism has been known to exist since antiquity; the Greek philosopher and mathematician Pythagoras was said to have warned his disciples against the dangers of eating fava beans (Scriver et al., 1995). The compounds vicine and isouramil, abundant in fava beans, are hypothesized to be the causative agents of the hemolytic response (Beutler, 1994).
Outside the areas where favism is prevalent, infection is probably the most common cause of hemolysis in subjects with G6PD deficiency. Oxidative metabolites produced by numerous bacterial, viral, and rickettsial infections have been identified as the cause of the anemic response. Particularly important infections that can precipitate a hemolytic episode are viral hepatitis, pneumonia, and typhoid fever (Cecil, 1992).
TREATMENT
Treatments for neonatal jaundice and hemolytic anemia
have existed for many years. These treatments insure that the body tissues will
be provided with enough oxygen by the red blood cells. Infants with prolonged
neonatal jaundice are placed under special lights, called bili-lights, which
alleviate the jaundice (Farid, personal interview). When an anemic episode
occurs, individuals are treated with nasal oxygen and are placed on bed rest,
which may afford symptomatic relief (Cecil, 1992). Anemic individuals are
sometimes treated with human haptoglobin products (Ohga et al., 1995), and/or
blood transfusions (Cecil, 1992). In acute hemolytic anemia, patients are
administered folic acid (Cecil, 1992).
Soon, G6PD deficient individuals will no longer have to worry about incurring a hemolytic episode in response to fava beans. Techniques are currently being developed to genetically engineer the fava bean so that the hypothesized causative agents of the hemolytic response are eliminated from the bean. This is especially significant since fava beans are an important part of the diet in the Middle East, where the frequency of G6PD deficiency and favism is high (see Figure 2).
Metabolic Disorders
Bernardo Haddock Lobo Goulart & Samanta Teixeira Basto
Medstudents' Homepage
Lysosomal Storage Diseases
1) Gaucher’s Disease
This condition is due to deficiency of glucosylceramidase, leading to a glucosylceramide lipidosis. There are infant, juvenile and adult forms. The infant form is characterized by early onset, marked hepatosplenomegaly and severe neurologic manifestations that result in early death. The juvenil form is similar to infant form, but neurologic manifestations are milder in the former.
The adult form, also called nonneuronopathic form, is the commonest lysosomal storage disease. The incidence is 30 times higher in ashkenazi jews ( 1 in 2500 births). Clinical manifestations include painless splenomegaly, which can lead to pancytopenia and bone pain of variable intensity. Patients may also present with pathological fractures, vertebral collapse and aseptic necrosis of femoral head. Diagnosis of bone disease may be done with MRI. Some degree, usually mild, of hepatic dysfuntion may occur, although severe hepatic failure, in rare cases, results in death. Characteristically, serum acid phosphatase is elevated. The disease is diagnosed by enzime assay, but the finding of a typical storage cell in bone marrow is almost diagnostic. This cell is also present in bone marrow of patients with granulocytic leukemia and multiple myeloma.
Treatment for the adult form includes partial or complete splenectomy, with posterior treatment of splenism. Bone marrow transplantation may be tried in life-threatening complications.
2) Fabry´s Disease
This condition reflects the accumulation of a trihexoside, galactosylgalactosylglucosylceramide, due to deficiency of a- galactosidase A. This disorder is X-linked and more common in men than women. Severe forms also occur in men and milder forms in women. Clinically, patients present with a painful neuropathy, mainly in the palms and soles, which can be intermittent or constant and shows a burning sensation. Painful abdominal crises may also occur, simulating an acute abdomen episode. Cutaneous manifestatons include angiokeratoma, characterized by angioectases that do not blanch with pressure, mainly in trunk, perineal area, penis and scrotum and hypohidrosis or anhidrosis, which can cause predispose to heat stroke with vigorous exercise. Occular findings, such corneal and lenticular opacities and tortuosity of retinal and conjunitival vessels are quite common. Cardiovascular manifestations result from lipid deposition on myocardium and are expressed by arrhytmias, acute myocardial infarction and orovalvular diseases. Small vessels involvement occur, predisposing patients to cerebral hemorrhages. Deposition of lipid in kidneys leads to progressive renal failure and this causes death in men at a median age of 41 years. Life expectancy is near normal in women.
Diagnosis is usually made through renal biopsy. Treatment includes counseling about the risks of anhidrosis, phenytoin for painful neuropathy and chronic dialysis for renal failure. Because donor´s kidney is not affected by the disease, renal transplantation is an acceptable alternative for the appropriate patient.
3) Niemann-Pick Disease :
This disease results from sphingomyelinase deficiency, constituting a sphingomyelin lipidosis. In type A, the most common form, onset is after birth, with hepatosplenomegaly, failure to thrive and neurologic impairment. Retinal cherry-red spots appear, but seizures and hipersplenism are uncommon manifestations. Diagnosis may be given by finding distinctive Niemann-Pick cells in bone marrow, but is necessary to confirm it with specific enzime assay. In type B, a relatively benign disorder occurs, with hepatosplenomegaly and, sometimes, pulmonary infiltrates. There are not neurologic signs. In type C, progressive neurologic deterioration occurs in childhood. This form is not due to sphingomyelinase deficiency, but is associated to massive lysosomal accumulation of cholesterol, reflecting an intracellular defect of cholesterol utilization.
Bibliography
BEAUDET, A.L. : Lysosomal Storage Disease, in Harrison´s Principles of Internal Medicine 12th ed, Mc Graw Hill, 1992.
METABOLIC DISORDERS
A. Acquired Disorders
Acquired
disorders associated with hypoxia are the most common. Anoxic poisons
(carbon monoxide, cyanide, carbon disulfide), hypoglycemia and ischemia produce
similar pathology in the nervous system.
1. Hypoxic Encephalopathy
- Hypoxic encephalopathy is characterized by necrosis of neurons in selectively vulnerable deep layers of cerebral cortex (laminar necrosis), Purkinje cells of cerebellum, hippocampal pyramidal cells and globus pallidus neurons.
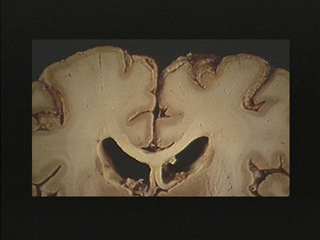
| Laminar necrosis in cortex is apparent in this section. |
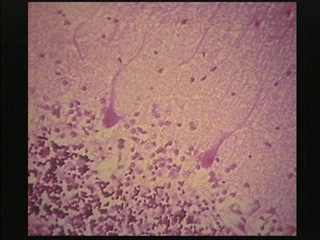
| This microscopic section from the cerebullar cortex shows loss of many Purbinyc cells & hypoxic change in the two present. |
2. Metabolic Encephalopathies
- Metabolic derangements secondary to renal or liver disease cause coma and relatively nonspecific morphologic changes in the neurons and/or glia.
3. Diabetes Mellitus
- Infants of diabetic mothers have huge hyperfunctioning Islets of Langerhans which overproduce insulin, and cause hypoglycemia and seizure. Diabetics may develop any of a wide gamut of neurological signs and neuropathological changes. Lesions at any level of central and peripheral nervous systems and muscle are probably secondary to vasa nervora as well as larger vessel disease. Secondary degeneration then occurs in peripheral somatic and visceral nerves and CNS, respectively.
FIGURE 3 The Pentose Phosphate Pathway. Note the importance of G6PD in the production of reduced G-SH, ribose, and NADPH (adapted from: Yoshida and Beutler, 1986, pg.8).
- NADP+ = nicotinamide adenine dinucleotide phosphate
- NADPH = reduced nicotinamide adenine dinucleotide phosphate
- GS-SG = oxidized glutathione
- G-SH = reduced glutathione
Table 2. 1990 criteria for the classification of polyarteritis nodosa (traditional format)*
- Criterion
- Definition
- 1. Weight loss =4 kg
- Loss of 4 kg or more of body weight since illness began, not due to dieting or other factors
- 2. Livedo reticularis
- Mottled reticular pattern over the skin of potions of the extremities or torso
- 3. Testicular pain or tenderness
- Pain or tenderness of the testicles, not due to infection, trauma, or other causes
- 4. Myalgias, weakness or leg tenderness
- Diffuse myalgias (excluding shoulder and hip girdle) or weakness of muscles or tenderness of leg muscles
- 5. Mononeuropathy or polyneuropathy
- Development of mononeuropathy, multiple mononeuropathys, or polyneuropathy
- 6. Diastolic BP >90 mm Hg
- Development of hypertension with diastolic BP higher than 90 mm Hg
- 7. Elevated BUN or creatinine
- Elevation of BUN >40 mg/dl or creatinine >1.5 mg/dl, not due to dehydration or obstruction
- 8. Hepatitis B virus
- Presenece of hepatitis B surface antigen or antibody in serum
- 9. Arteriographic abnormality
- Arteriogram showing aneurysms or occlusions of the visceral arteries, not due to arteriosclerosis, fibromuscular dysplasia, or other noninflammatory causes
- 10. Biopsy of small or medium-sized artery containing PMN
- Histologic changes showing the presence of granulocytes or granulocytes and mononuclear leukocytes in the artery wall
* For classification purposes, a patient shall be said to have polyarteritis nodosa if at least 3 of these 10 criteria are present. The presence of any 3 or more criteria yields a sensitivity of 82.2% and a specificicy of 86.6%. BP = blood pressure; BUN = blood urea nitrogen; PMN = polymorphonuclear neutrophils.
| Debrancher Enzyme Deficiency
(DBD) (Also known as Cori's or Forbes' Disease) A Guide to Related Materials on MDA's Web Site | |
| Quick Definition:
Onset · Early childhood in first year. Symptoms · Generalized weakness and muscle wasting. Enlarged liver in infancy. Episodes of low blood sugar. Progression · Slow to variable progression. Muscular symptoms may be delayed until early teens and adulthood. Inheritance · Autosomal recessive. | |
|
|
Glycogen Storage Disease Type I
glucose-6-phosphatase deficiency, Von Gierke disease)
Children with GSD I are unable to release glucose from liver glycogen. If untreated this results in prolonged periods when their blood sugar level is too low. They become unwell in early childhood with sweating, irritability, poor growth and muscle weaknes s. their livers become enlarged because of excessive accumulation of glycogen that cannot be broken down normally. In addition to these problems, children with GSD I can develop frequent mouth ulcers and are at increased risk of infection. Treatment primar ily consists of giving glucose drinks frequently during the day and, in most cases, continuously overnight through a tube passed down the nose into the stomach (a nasogastric tube). As children get older, treatment with cornstarch, which releases glucose sl owly into the gut, may be very effective. With such intensive treatment most children do well and their symptoms improve as they reach adulthood.
Glycogen Storage Disease type II
alpha glucosidase (acid maltase) deficiency, Pompe's Disease
GSD II usually presents within the first months of life with severe muscle weakness and heart muscle involvement. Unfortunately, no treatment has been found to prevent the progression of the most severe (infantile) form of this disorder and affected childre n die from heart failure, usually before the age of 18 months. There are however, milder forms of GSD II in which the heart is not affected and where symptoms do not develop until later in childhood or in adult life and the progression of the illness is slo wer. Some individuals in this latter category have improved with a special high protein diet
Glycogen Storage Disease type III
debrancher enzyme deficiency, Cori disease
Children with GSD III are often first diagnosed because they have been noticed to have a swollen abdomen due to a very large liver. Some children have problems with low blood sugars on fasting but this is not as common as in GSD I. Growth may be delayed during childhood but the majority attain a normal final adult height. Although some older individuals develop problems with muscle weakness (which may also affect the heart) the outcome for most is good with the liver returning t o a normal size with age. Treatment consists of a high protein diet and prevention of prolonged periods of fasting.
<A HREF="agsdhome.html>Back to the AGSD home page
Glycogen Storage Disease type IV
brancher enzyme deficiency, Anderson disease
GSD IV is a very severe but rare disorder that leads to cirrhosis of the liver and heart involvement. Most children with this condition have died before two years of age. No treatment apart from liver transplantation has been found to prevent progression o f the disease.
Glycogen Storage Disease type V
muscle glycogen phosphorylase deficiency, McArdle disease
Glycogen Storage Disease type VII
muscle phosphofructokinase deficiency, Tauri disease
GSD V and GSD VII affect muscle tissue only. Symptoms include painful muscle cramps during exercise and muscle weakness. Treatment primarily consists of avoiding strenuous exercise which, as well as causing pain, may lead to kidney damage. Some patients ha ve been helped by a high protein diet.
Glycogen Storage Disease type VI
liver phosphorylase deficiency, Hers disease
Glycogen Storage Disease type IX
liver glycogen phosphorylase kinase deficiency
GSD VI and GSD IX are two of the least severe forms of GSD. In most individuals apart from liver enlargement there are few other problems. There is usually no tendency to low blood sugar, the liver becomes smaller with age and children grow normally. Unlike other forms of GSD, most cases of GSD IX affect males (X-linked inheritance).
What is Glycogen Storage Disease (GSD)?
Glucose is a major source of energy for the body. It is stored in the form of glycogen in both the liver and muscles and later released with the help of enzymes. Persons affected by GSD have an inherited defect in one of the enzymes responsible for forming or releasing glycogen as it is needed by the body during exercise and/or between meals. There are about eleven known types of GSD which are classified by a number, by the name of the defective enzyme, or by the name of the doctor who first described the condition. For example, GSD I, a defect in the enzyme glucose-6-phosphatase, was originally known as von Gierke's Disease.
GSD can affect the liver, the muscles or both. Diagnosis of the type of GSD is made on the basis of an individual's symptoms, the results of a physical examination and of biochemical tests. Occasionally, a muscle or liver biopsy is required to confirm the a ctual enzyme defect. All forms of GSD, except some forms of the liver phosphorylase kinase deficiency (GSD IX), occur when a child inherits the affected gene from both parents (autosomal recessive inheritance) each of whom is a carrier but not affected them selves. This means that for each pregnancy there is a 1 in 4 chance that the child will inherit both defective genes and thereby be affected.
Types of Glycogen Storage Disease
Some forms of GSD cause little in the way of illness, while others are life-threatening. Click on the links below for a description of the general symptoms, current treatment and long-term outcome for the most commonly diagnosed glycogen storage diseas es.
GSD I (glucose-6-phosphatase deficiency, Von Gierke disease)
GSD II (alpha glucosidase deficiency, Pompe's disease). There is also a longer document on Pompe's disease which explains in simple terms the background behind the disease and current research taking place into developing a treatment.
GSD III (debrancher enzyme deficiency, Cori disease)
GSD IV (brancher enzyme deficiency, Anderson disease)
GSD V (muscle glycogen phosphorylase deficiency, McArdle disease) and GSD VII (muscle phosphofructokinase deficiency, Tauri disease)
GSD VI (liver phosphorylase deficiency, Hers disease) and GSD IX (liver glycogen phosphorylase kinase deficiency)
Glycogen Storage Disease Type 1A
Synonyms Von Gierke disease. Hepatorenal GSD
McKusick No. 23220
1. Physical
2. Neurological
3. Gastrointestinal
4. Renal
5. Bone
6. Comment
7. Haematological
8. Biochemical
Enzyme Glucose-6-phosphatase
EC number 3.1.3.9
................Diagnostics and therapy................
Treatment
Carrier detection
Prenatal diagnosis
Glycogen Storage Disease Type II [Infantile]
Synonyms Pompe disease, Acid Maltase Deficiency
McKusick No. 23230
1. Physical
2. Neurological
3. Gastrointestinal
4. Renal
5. Bone
6. Comment
7. Haematological
8. Biochemical
EC number
................Diagnostics and therapy................
Treatment
Carrier detection
Prenatal diagnosis
Glycogen Storage Disease Type III
Synonyms Forbes, Cori Disease; Limit Dextrinosis
McKusick No. 23240
1. Physical
2. Neurological
3. Gastrointestinal
4. Renal
5. Bone
6. Comment
7. Haematological
8. Biochemical
Enzyme Debrancher enzyme, amylo-1,6-glucosidase
EC number 3.2.1.33
................Diagnostics and therapy................
Treatment
Carrier detection
Prenatal diagnosis
Inherited conditions in man
-
- AT types A & C Full account (OMIM)
- AT type D Full account (OMIM)
- AT with skin pigmentation Full account (OMIM)
- AT-like syndrome Full account (OMIM)
-
- Bloom syndrome Full account (OMIM)
-
- see muco-polysaccaride diseases
-
- A case study - and more
- Duchenne de Boulogne - who was he?
Familial Mediterranean fever
-
- see muco-polysaccaride diseases
-
- see muco-polysaccaride diseases
-
- Huntingdon disease - a handbook.
-
- see also muco-polysaccaride diseases
- Basic deatails
-
- Support group home page
- What is it?
-
- see muco-polysaccaride diseases
-
- see muco-polysaccaride diseases
-
- General account (Canadian)
Pelizaeus-Merzbacher
Retinal problems
-
- Macular degeneration Best's disease
- Macular degeneration - another form
- Detachment Stickler syndrome
-
- see muco-polysaccaride diseases
-
- A Family Handbook - very large.
- Risperidone - drug treatment
- Current basic science
- Medications
- Managing the illness
- Questions & answers
- Pineal gland involvement
-
- see muco-polysaccaride diseases
Sly disease
-
- see muco-polysaccaride diseases
-
- Werner syndrome Full account (OMIM)
Other general conditions
Inherited disorders of the kidney
| Duchenne Muscular Dystrophy (DMD) (Also known as Pseudohypertrophic) [read more] | ||||||||||
| Onset | Early childhood - about 2 to 6 years. | |||||||||
| Symptoms | Generalized weakness and muscle wasting affecting limb and trunk muscles first. Calves often enlarged. | |||||||||
| Progression | Disease progresses slowly but will affect all voluntary muscles. Survival rare beyond late twenties. | |||||||||
| Inheritance | X-linked recessive (females are carriers). | |||||||||
| Becker Muscular Dystrophy (BMD) [read more] | ||||||||||
| Onset | Adolescence or adulthood. | |||||||||
| Symptoms | Almost identical to Duchenne but often much less severe. Can be significant heart involvements. | |||||||||
| Progression | Slower and more variable than Duchenne with survival well into mid to late adulthood. | |||||||||
| Inheritance | X-linked recessive (females are carriers). | |||||||||
| Emery-Dreifuss Muscular Dystrophy (EDMD) [read more] | ||||||||||
| Onset | Childhood to early teens. | |||||||||
| Symptoms | Weakness and wasting of shoulder, upper arm and shin muscles. Joint deformities are common. | |||||||||
| Progression | Disease usually progresses slowly. Frequent cardiac complications are common. | |||||||||
| Inheritance | X-linked recessive (females are carriers). | |||||||||
| Limb-Girdle Muscular Dystrophy (LGMD) [read more] | ||||||||||
| Onset | Late childhood to middle age. | |||||||||
| Symptoms | Weakness and wasting affecting shoulder and pelvic girdles first. | |||||||||
| Progression | Usually progresses slowly with cardiopulmonary complications often occurring in later stages of the disease. | |||||||||
| Inheritance | Autosomal recessive, X-linked recessive. | |||||||||
| Facioscapulohumeral Muscular Dystrophy (FSH or FSHD) (Also known as Landouzy-Dejerine) [read more] | ||||||||||
| Onset | Childhood to early adulthood. | |||||||||
| Symptoms | Facial muscle weakness, with weakness and wasting of the shoulders and upper arms. | |||||||||
| Progression | Progresses slowly with some periods of rapid deterioration. Disease may span many decades. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
| Myotonic Dystrophy (DM) (Also known as Steinert's Disease) [read more] | ||||||||||
| Onset | Childhood to middle age. | |||||||||
| Symptoms | Generalized weakness and muscle wasting affecting face, feet, hands and neck first. Delayed relaxation of muscles after contraction. Congenital myotonic form is more severe. | |||||||||
| Progression | Progression is slow, sometimes spanning 50 to 60 years. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
| Oculopharyngeal Muscular Dystrophy (OPMD) [read more] | ||||||||||
| Onset | Early adulthood to middle age. | |||||||||
| Symptoms | First affects muscles of eyelid and throat. | |||||||||
| Progression | Slow progression with swallowing problems common as disease progresses. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
| Distal Muscular Dystrophy (DD) [read more] | ||||||||||
| Onset | 40-60 years. | |||||||||
| Symptoms | Weakness and wasting of muscles of the hands, forearms and lower legs. | |||||||||
| Progression | Slow progression but not life-threatening. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
| Congenital Muscular Dystrophy (CMD) [read more] | ||||||||||
| Onset | At birth. | |||||||||
| Symptoms | Generalized muscle weakness with possible joint deformities. | |||||||||
| Progression | Disease progresses very slowly. Fukuyama form is more severe and involves mental functions. | |||||||||
| Inheritance | Autosomal recessive, autosomal dominant. | |||||||||
MOTOR NEURON DISEASES: |
||||||||||
| Amyotrophic Lateral Sclerosis (ALS) (Also known as Lou Gehrig's Disease) [read more] | ||||||||||
| Onset | Adulthood. | |||||||||
| Symptoms | Generalized weakness and muscle wasting with cramps and muscle twitches common. | |||||||||
| Progression | ALS first affects legs, arms and/or throat muscles. Usually progresses rapidly with 3 to 5 year average survival. | |||||||||
| Inheritance | Autosomal dominant, autosomal recessive. | |||||||||
| Infantile Progressive Spinal Muscular Atrophy (SMA, SMA1 or WH) (Also known as SMA Type 1, Werdnig-Hoffman) [read more] | ||||||||||
| Onset | Before birth to 3 months. | |||||||||
| Symptoms | Generalized muscle weakness, weak cry, trouble swallowing as well as sucking, and breathing distress. Cannot sit up. | |||||||||
| Progression | Progresses very rapidly with early childhood death. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Intermediate Spinal Muscular Atrophy (SMA or SMA2) (Also known as SMA Type 2) [read more] | ||||||||||
| Onset | 6 months to 3 years. | |||||||||
| Symptoms | Weakness in arms, legs, upper and lower torso, often with skeletal deformities. | |||||||||
| Progression | Disease usually progresses rapidly and respiratory problems may develop. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Juvenile Spinal Muscular Atrophy (SMA, SMA3 or KW) (Also known as SMA Type 3, Kugelberg-Welander) [read more] | ||||||||||
| Onset | 1 to 15 years. | |||||||||
| Symptoms | Weakness in leg, hip, shoulder, arm and sometimes respiratory muscles. | |||||||||
| Progression | Disease progresses slowly. Wheelchair often required later in life. Life span usually not affected. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Spinal Bulbar Muscular Atrophy (SBMA) (Also known as Kennedy's Disease and X-Linked SBMA) [read more] | ||||||||||
| Onset | Adulthood (20 to 50 years - variable severity). | |||||||||
| Symptoms | Weakness and muscle wasting of bulbar region (mouth and throat) and skeletal muscles. Usually affects only men -- women as carriers may have a mild form. Facial fasciculations and mild sensory involvement are common. | |||||||||
| Progression | Slow, variable progression, sometimes accompanied by breast development, infertility and testicular wasting in men. Normal life span. | |||||||||
| Inheritance | X-linked recessive (females are carriers). | |||||||||
| Adult Spinal Muscular Atrophy (SMA) [read more] | ||||||||||
| Onset | 18 to 50 years. | |||||||||
| Symptoms | Generalized weakness and muscle wasting with muscle twitches common. X-linked form affects men only and involves muscles of mouth and throat as well as other muscles. | |||||||||
| Progression | Variable disease progression. Relatively mild form of SMA with little impact on life expectancy. | |||||||||
| Inheritance | Autosomal dominant, autosomal recessive. | |||||||||
INFLAMMATORY MYOPATHIES: | ||||||||||
| Dermatomyositis (PM/DM) [read more] | ||||||||||
| Onset | Childhood to late adulthood. | |||||||||
| Symptoms | Weakness of neck and limb muscles. Muscle pain and swelling common. Skin rash typically affecting cheeks, eyelids, neck, chest and limbs. | |||||||||
| Progression | Disease progression and severity vary among individuals. Often responds to drug therapy. | |||||||||
| Polymyositis (PM/DM) [read more] | ||||||||||
| Onset | Childhood to late adulthood. | |||||||||
| Symptoms | Weakness of neck and limb muscles. Muscle pain and swelling common. Sometimes associated with malignancy. | |||||||||
| Progression | Disease severity and progression vary among individuals. Often responds to drug therapy. | |||||||||
DISEASES OF THE NEUROMUSCULAR JUNCTION: | ||||||||||
| Myasthenia Gravis (MG) [read more] | ||||||||||
| Onset | Childhood to adulthood. | |||||||||
| Symptoms | Weakness and fatigability of muscles of eyes, face, neck, throat, limbs and/or trunk. | |||||||||
| Progression | Disease progression varies. Drug therapy and/or removal of thymus gland often effective. | |||||||||
| Lambert-Eaton Syndrome (LES) [read more] | ||||||||||
| Onset | Adulthood to middle age. | |||||||||
| Symptoms | Weakness and fatigue of hip muscles with aching back and thigh muscles common. Lung tumor is sometimes present. | |||||||||
| Progression | Progression varies with success of drug therapy and treatment of any malignancy. | |||||||||
MYOPATHIES DUE TO ENDOCRINE ABNORMALITIES: | ||||||||||
| Hyperthyroid Myopathy (HYPTM) [read more] | ||||||||||
| Onset | Childhood to adulthood. | |||||||||
| Symptoms | Weakness of upper arm and upper leg muscles. Some muscle wasting. | |||||||||
| Progression | Usually improves with treatment of underlying thyroid condition. | |||||||||
| Hypothyroid Myopathy (HYPOTM) [read more] | ||||||||||
| Onset | Childhood to adulthood. | |||||||||
| Symptoms | Weakness of arm and leg muscles. Stiffness and muscle cramps common. | |||||||||
| Progression | Usually improves with treatment of underlying thyroid condition. | |||||||||
DISEASES OF PERIPHERAL NERVE: | ||||||||||
| Charcot-Marie-Tooth Disease (CMT) (Also known as Hereditary Motor and Sensory Neuropathy (HMSN) or Peroneal Muscular Atrophy (PMA)) [read more] | ||||||||||
| Onset | Childhood to young adulthood. | |||||||||
| Symptoms | Weakness and atrophy of muscles of hands and lower legs, with foot deformities and some loss of sensation in feet. | |||||||||
| Progression | Slow but variable progression among individuals. Normal life span. | |||||||||
| Inheritance | Autosomal dominant, autosomal recessive, X-linked recessive, X-linked dominant. | |||||||||
| Dejerine-Sottas Disease (DS) (Also known as CMT Type 3 or Progressive Hypertrophic Interstitial Neuropathy) [read more] | ||||||||||
| Onset | Early childhood. | |||||||||
| Symptoms | Same as CMT, but more severe. Delayed motor development in childhood. Weakness and muscle wasting affecting hands and lower legs. Variable loss of sensation in feet. | |||||||||
| Progression | Severity and progression of disease vary. | |||||||||
| Inheritance | Believed to be autosomal dominant. | |||||||||
| Friedreich's Ataxia (FA) [read more] | ||||||||||
| Onset | Childhood to adolescence. | |||||||||
| Symptoms | Impairment of limb coordination, with weakness and muscle wasting. | |||||||||
| Progression | Severity and progression of disorder vary. Often associated with diabetes/heart disease. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
OTHER MYOPATHIES: | ||||||||||
| Myotonia Congenita (MC) (Two forms: Thomsen's and Becker's Disease) [read more] | ||||||||||
| Onset | Infancy to childhood. | |||||||||
| Symptoms | Muscle stiffness and cramps usually occurring after periods of rest. With activity, returns to normal muscle function. | |||||||||
| Progression | Condition causes discomfort but is not life-threatening. | |||||||||
| Inheritance | Autosomal dominant, autosomal recessive. | |||||||||
| Paramyotonia Congenita (PC) [read more] | ||||||||||
| Onset | Childhood to early adulthood. | |||||||||
| Symptoms | Poor or difficult relaxation of muscles, which may worsen after repeated use or exercise. Often may be associated with hyperkalemic periodic paralysis. | |||||||||
| Progression | Condition causes discomfort throughout life but is not life-threatening. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
| Central Core Disease (CCD) [read more] | ||||||||||
| Onset | Early infancy to childhood. | |||||||||
| Symptoms | Delayed motor development. Hip displacement at birth not uncommon. | |||||||||
| Progression | Variable severity and progression. May be disabling. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
| Nemaline Myopathy (NM) [read more] | ||||||||||
| Onset | Early childhood. | |||||||||
| Symptoms | Delayed motor development. Weakness of arm, leg, trunk, face and throat muscles. | |||||||||
| Progression | Severity and progression vary. Life expectancy is threatened. | |||||||||
| Inheritance | Autosomal dominant, autosomal recessive. | |||||||||
| Myotubular Myopathy (MTM or MM) [read more] | ||||||||||
| Onset | Infancy. | |||||||||
| Symptoms | Drooping of upper eyelids, facial weakness, blackout spells. Weakness of the limbs and trunk muscles. Reflexes usually absent. | |||||||||
| Progression | Slow progression. | |||||||||
| Inheritance | X-linked recessive, autosomal recessive, autosomal dominant. | |||||||||
| Periodic Paralysis (PP) (Two forms: Hypokalemic - HYPOP - and Hyperkalemic - HYPP) [read more] | ||||||||||
| Onset | Childhood to adulthood. | |||||||||
| Symptoms | Episodes of generalized muscle weakness with periods of paralysis affecting arms, legs and neck. Hyperkalemic type may be associated with paramyotonia congenita. | |||||||||
| Progression | Frequency of attacks and severity vary. May respond to drug therapy. | |||||||||
| Inheritance | Autosomal dominant. | |||||||||
METABOLIC DISEASES OF MUSCLE: | ||||||||||
| Phosphorylase Deficiency (MPD or PYGM) (Also known as McArdle's Disease) [read more] | ||||||||||
| Onset | Childhood to adolescence. | |||||||||
| Symptoms | Muscle cramps usually occurring after exercise. Intense exercise can cause muscle destruction and possible kidney damage. | |||||||||
| Progression | Variable severity and progression. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Acid Maltase Deficiency (AMD) (Also known as Pompe's Disease) [read more] | ||||||||||
| Onset | Infancy to adulthood. | |||||||||
| Symptoms | In infant form, disease is generalized and severe, with heart, liver and tongue enlargement common. Adult form involves weakness of upper arms and legs, trunk and respiratory muscles. | |||||||||
| Progression | Progression varies. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Phosphofructokinase Deficiency (PFKM) (Also known as Tarui's Disease) [read more] | ||||||||||
| Onset | Childhood. | |||||||||
| Symptoms | Muscle fatigue that, upon exercise, can lead to severe cramps, nausea, vomiting, muscle damage and discoloration of urine. | |||||||||
| Progression | Progression varies widely. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Debrancher Enzyme Deficiency (DBD) (Also known as Cori's or Forbes' Disease) [read more] | ||||||||||
| Onset | Early childhood in first year. | |||||||||
| Symptoms | Generalized weakness and muscle wasting. Enlarged liver in infancy. Episodes of low blood sugar. | |||||||||
| Progression | Slow to variable progression. Muscular symptoms may be delayed until early teens and adulthood. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Mitochondrial Myopathy (MITO) [read more] | ||||||||||
| Onset | Early infancy to adulthood. | |||||||||
| Symptoms | Generalized muscle weakness, flaccid neck muscles and inability to walk. Brain is often involved, with seizures, deafness, loss of balance and vision, and retardation common. | |||||||||
| Progression | Wide variety of progression and severity. | |||||||||
| Inheritance | Maternal mitochondrial gene (mtDNA). | |||||||||
| Carnitine Deficiency (CD) [read more] | ||||||||||
| Onset | Early childhood. | |||||||||
| Symptoms | Varied weakness of shoulders, hips, face and neck muscles. | |||||||||
| Progression | Progression varies and carnitine supplementation is often effective. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Carnitine Palmityl Transferase Deficiency (CPT) [read more] | ||||||||||
| Onset | Young adulthood. | |||||||||
| Symptoms | Inability to sustain moderate prolonged exercise. Prolonged exercise and/or fasting can cause severe muscle destruction with urine discoloration and kidney damage. | |||||||||
| Progression | Severity varies. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Phosphoglycerate Kinase Deficiency (PGK) [read more] | ||||||||||
| Onset | Childhood to adolescence. | |||||||||
| Symptoms | Muscle pain and weakness, with muscle damage and urine discoloration possible after vigorous exercise. | |||||||||
| Progression | Severity varies. Avoid intense exercise. | |||||||||
| Inheritance | X-linked recessive, autosomal recessive. | |||||||||
| Phosphoglycerate Mutase Deficiency (PGAM or PGAMM) [read more] | ||||||||||
| Onset | Childhood to adulthood. | |||||||||
| Symptoms | Muscle pain, cramps, muscle damage and urine discoloration possible during intense exercise of brief duration. | |||||||||
| Progression | Severity varies. Avoid intense exercise. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Lactate Dehydrogenase Deficiency (LDHA) [read more] | ||||||||||
| Onset | Childhood to adolescence. | |||||||||
| Symptoms | Exercise intolerance with muscle damage and urine discoloration possible following strenuous physical activity. | |||||||||
| Progression | Severity varies. Avoid intense exercise. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
| Myoadenylate Deaminase Deficiency (MAD) [read more] | ||||||||||
| Onset | Early adulthood to middle age. | |||||||||
| Symptoms | Muscle fatigue and weakness during and after exertion, with muscle soreness or cramping. May not attain prior performance levels. | |||||||||
| Progression | Severity varies. Usually nonprogressive and non-debilitating. | |||||||||
| Inheritance | Autosomal recessive. | |||||||||
Von Gierke Disease
Synonyms
It is possible that Von
Gierke Disease may not be the name that you expected. Your physician may have
given you another name for this disease. Please check the synonyms listed below
to find other names for this specific disorder.
Glycogen Storage Disease I
Glycogenosis Type I
Hepatorenal
Glycogenosis
Disorder
Subdivisions: ![]()
Glycogenosis Type IA
Glucose-G-Phosphatase
Deficiency
Glycogenosis Type IB
Glucose-6-Phospate Translocase
Deficiency
Glucose-6-Phosphate Tranport Defect
Abstract (General Discussion) ![]()
The information contained in the Rare Disease Database (RDB) is
provided for educational purposes only. It should not be used for diagnostic or
treatment purposes. If you order the full text version of this report from NORD,
you can contact the agencies listed in the Resources section for more detailed
information and avenues to support. In addition, your personal physician may be
able to provide details specific to your case.
Von Gierke Disease is a glycogen storage disease. This hereditary metabolic
disorder is caused by an inborn lack of the enzyme glucose-6-phosphatase. This
enzyme is needed to convert the main carbohydrate storage material (glycogen)
into sugar (glucose), which the body uses for its energy needs. A deficiency
causes deposits of excess glycogen in the liver and kidney cells.
ADRENAL TESTING
Ed Friedlander, M.D., Pathologist
INTRODUCTION
-
- the thin, tired patient who has no appetite: Addison's disease?
-
- the obese patient who may be :
may have high blood pressure,
may have hyperglycemia, and may have many other problems: Cushing's syndrome? (End. Metab. Clin. N.A. 17(3): 445, 1988; Mayo Clin. Proc. 61: 49, 1988; Am. J. Clin. Path. 90: 345, 1988.)
(order serum cortisol determinations at 8 AM and 4-8 PM, plus a low-dose dexamethasone suppression test, or order a 24 hr urinary free cortisol)
-
- the patient with high blood pressure and low serum potassium: primary aldosteronism (Conn's syndrome?)
-
- the nervous patient with high blood pressure and headaches: pheochromocytoma?
-
- the kid with just about any puzzling problem?
Because a single laboratory test seldom establishes the final diagnosis in adrenal disease, you must know what you are doing!
If you fail to diagnose a disease which is present, the patient is likely to die of a disease which would have responded well to treatment. (Suicide is common among patients with Cushing's syndrome.)
If you make the correct diagnosis, the treatment of most of these diseases is very satisfying to physician and patient alike.
The whole business is very complicated. If your screening tests support your idea that the patient has any endocrine disease, it is usually best to obtain consultation with an endocrinologist.
ADRENAL CORTICAL DISEASES (all about kind of testing: Mayo Clin. Proc. 67: 1055, 1992; Cushingism Ann. Int. Med. 112: 434, 1990 and Postgrad. Med. 86(8): 79, Dec. 1989)
Cushing's disease (pituitary adenoma or microadenoma): increased ACTH
Adrenal cortical adenoma or carcinoma: increased ACTH (maybe....)
Ectopic ACTH production (i.e., oat cell carcinoma, carcinoid, thymoma, pheochromocytoma) or CRF production (oat cell, rarely others)
Iatrogenic (glucocorticoid therapy)
Addison's disease: decreased cortisol (or decreased ability to produce cortisol)
Primary addisonism (autoimmune, TB, after steroid Rx, etc.): increasedincreased ACTH
Secondary addisonism (hypopituitarism): decreased ACTH (maybe....)
Hyperaldosteronism: increased aldosterone (maybe); increased aldosterone/renin ratio (i.e., >250 or >980 or whatever's your cutoff)
Secondary (congestive heart failure, cirrhosis, etc.)
"Congenital adrenal hyperplasia" (virilization syndromes, etc. Once considered rare, mild forms are now recognized as among the most common illnesses.
PLASMA ACTH BY RADIOIMMUNOASSAY (normal 60 pg/mL at 9 AM)
ACTH is very susceptible to plasma proteases after the specimen is drawn, and tends to stick to glassware. The assay is expensive, not very sensitive, the plasma must be drawn into an iced purple-top tube and frozen rapidly, etc. etc.
One time when you probably should order a serum ACTH is when your patient has proved Cushingism, and you have ruled out Cushing's disease (see below), though you will not want to delay the workup if the test needs to be sent out.
Low ACTH indicates adrenal cortical adenoma or hyperplasia. Nice algorithm: Mayo. Clin. 61: 49, 1988.
ACTH levels are also quite helpful after hypophysectomy for Cushing's disease (return of circadian rhythm following adequate treatment), and in the diagnosis of ectopic ACTH syndrome (very high levels).
PLASMA CORTISOL BY RADIOIMMUNOASSAY
Estrogens (oral contraceptive pill, pregnancy, etc.) increase transcortin levels and thus "total cortisol".
*This commonly-performed assay is replacing three obsolete tests.
-
- cortisol ("hydrocortisone")
- cortisone (exogenous, probably)
- 11-deoxycortisol
Serum cortisol by fluorimetric assay ("Mattingly")
Competitive protein binding: an old-fashioned radioassay.
Plasma cortisol is generally increased in Cushing's syndrome, and loss of diurnal variation (* 4 PM cortisol more than half 8 AM value) suggests Cushingism.
A "spot" plasma cortisol may be normal in all but the worst cases of adrenal insufficiency (addisonism). Therefore, it should not be used alone as a screening test for adrenal insufficiency! (See Pharmacotherapy 9: 269, 1989.)
URINARY FREE CORTISOL (normal 24-108 micrograms/24 hr)
At plasma cortisol concentrations above about 25 mcg/dL, transcortin becomes saturated and unbound, unconjugated cortisol spills into the glomerular filtrate.
Normal urinary free cortisol ranges around 20-90 mcg/24 hr. Of course, urinary free cortisol, unlike plasma cortisol, gives an integrated value over time.
This test is worthless for the detection or differential diagnosis of adrenal insufficiency.
* URINARY 17-OHCS (17-HYDROXYCORTICOSTEROIDS, PORTER-SILBER CHROMOGENS)
-
- tetrahydro-cortisol glucuronide
- tetrahydro-cortisone glucuronide
- tetrahydro-11-deoxycortisol (may be removed with CCl4)
Normal range is around 4-15 mg/24 hr or (better) 3-7 mg/gm creatinine.
URINARY 17-KS (17-KETOSTEROIDS)
-
- *dehydroepiandrosterone (DHEA)
- *androsterone sulfate
- *etiocholanolone sulfate
*These substances react with meta-dinitrobenzene to produce colored compounds (the "Zimmermann reaction").
*Normal range is around 7-20 mg/24 hr or (better) 4-10 mg/gm creatinine. We do not measure these in serum.
*Fractionation may aid in the diagnosis of adrenal cortical carcinoma (increased dehydroepiandrosterone, a beta-ketosteroid), and of inborn errors of metabolism.
* URINARY 17-KGS (17-KETOGENIC STEROIDS)
This test is not specific for much of anything, and not in widespread use any more.
ACTH STIMULATION TEST (B.M.J. 298: 271, 1989)
One rapid test to screen for Addison's disease requires synthetic ACTH ("Cortrosyn", "tetracosactrin", "cosyntropin" etc., 250 mcg I.M. or IV), with plasma cortisol measured just before and one hour after.
In Addison's disease, there is little or no response. You must confirm the diagnosis with a prolonged infusion of ACTH. See Am. J. Med. 79: 679, 1985.
One prolonged infusion test requires synthetic ACTH 500 mcg to be given over 8 hrs each day for one to five days, with plasma cortisol measured before, during, and after each infusion, and urinary hormones measured daily.
In a patient with primary adrenal insufficiency, there is little or no response on the first day. There may be a slight response on the second and third days, but this is likely to taper off on the fourth and fifth days. (Why?)
In a patient with adrenal insufficiency secondary to pituitary insufficiency (low ACTH), there will be a greater response on each successive day the test is run ("staircase"). (Remember to check these patients for deficiencies in other anterior pituitary hormones!)
*The test is also occasionally used in suspected Cushingism, though this is much less useful.
In Cushing's syndrome caused by an autonomously secreting adrenal cortical adenoma, there may or may not be a response to ACTH infusion. (Carcinomas will almost never respond.)
In hypokalemic alkalosis (rarely overt Cushingism) caused by ectopic ACTH secretion by a lung cancer, etc., there may or may not be a response to ACTH infusion.
*The old (but interesting) Thorn Test involved giving an injection of ACTH and observing its effect on the absolute eosinophil count. Failure of the "eo" count to drop by over 50% indicated Addison's disease.
For the use of an ACTH stimulation test to diagnose congenital adrenal hyperplasia, see below.
Warnings:
When you suspect adrenal insufficiency, administer dexamethasone 2 mg before the procedure begins. This will prevent Addisonian crisis. (* It also prevented the frequent allergic responses to the bovine ACTH that was once used for the test.)
METYRAPONE TEST
Metyrapone ("Metopirone") is a potent inhibitor of 11-hydroxylation (* etc.).
One of the newer versions requires metyrapone 30 mg/kg at bedtime, and morning plasma for 11-deoxycortisol and ACTH.
The standard oral test for an adult requires metyrapone 750 mg q 4 hr x 6 doses. (Contraindicated in renal failure!)
In patients with adrenal insufficiency with some adrenal reserve, this test can demonstrate a component of pituitary insufficiency. See Arch. Int. Med. 143: 2276, 1983.
In Cushing's disease (i.e., a demonstrable or presumed pituitary adenoma making ACTH), there is usually an exaggerated response (3-6 fold increase in urinary 17-OHCS, etc.)
In Cushing's syndrome caused by an autonomously secreting adrenal cortical adenoma or carcinoma, response is usually diminished or absent (i.e., the ACTH-producing cells of the anterior pituitary have been chronically suppressed.)
In the syndrome of ectopic ACTH production, there may or may not be a response to metyrapone.
The main use of the metyrapone test nowadays is to distinguish Cushing's disease from other Cushing syndromes.
OTHER WAYS OF TESTING PITUITARY ACTH PRODUCTION
Its use in distinguishing Cushing's disease from other causes of Cushingism has been superseded by safer tests.
The corticotropin-releasing factor stimulation test measures the response to a dose of this substance by patients with Cushing's syndrome.
Patients with the ectopic ACTH syndrome or adrenal cortical tumors show no response to CRF (NEJM 310: 622, 1984).
The CRF stimulation test is now considered a good complement to the high-dose dexamethasone test (see below) for Cushing's disease. Both are about 90% accurate (Lancet 2: 540, 1986; Ann. Int. Med. 105: 682, 1986). The current trend seems to favor it over the high-dose dexamethasone test (JAMA 269: 2232, 1993; a new version which adds a bit of dexamethasone during the test).
DEXAMETHASONE SUPPRESSION TESTS
In Cushing's disease, the pituitary gland loses some of its sensitivity to feedback inhibition, so there will be little or no decrease in ACTH or cortisol unless the dose of dexamethasone is very high.
Adrenal cortical tumors that overproduce glucocorticoids are not under the control of ACTH (which is suppressed anyway!) and there will be no response to any dose of dexamethasone.
Low dose dexamethasone suppression tests are used to rule out Cushing's syndrome.
Ten percent of patients without Cushing's syndrome will still flunk this test ("pseudo-Cushingism"). This includes patients who:
-
- are obese (at least the books say this, though the Mayo Clinic group disagrees. Fat people are often worked up for "glands".)
- are depressed (especially if it's so bad they aren't eating or sleeping. Ask a psychiatrist about the usefulness of this test in the diagnosis of various types of depression. Its use by general internists is discouraged. See Arch. Int. Med. 143: 2085, 1983. This test cannot distinguish very depressed patients from those with Cushing's disease.)
- are alcoholics who are still drinking, perhaps secretly
- are taking estrogens, or are pregnant (increased serum transcortin)
- are taking phenytoin, which speeds the metabolism of dexamethasone (and metyrapone too....)
PRIMARY ALDOSTERONISM (End. Metab. Clin. N.A. 17(2): 367, 1988; Mayo Clin. 65: 96, 1990).
The following workup is very complex, and requires documenting inappropriate hyperkaliuria, difficulty to stimulate renin, and inability to suppress aldosterone.
The best news lately in adrenal testing is that a high plasma aldosterone/renin level, drawn at 8 AM after a two-hour walk provided complete separation of patients with primary hyperaldosteronism from patients with essential hypertension or normal blood pressure. Further, it distinguished adenoma patients from patients with idiopathic adrenal hyperplasia (see below). Reference Arch. Int. Med. 153: 2125, 1993; also J. Clin. End. Metab. 73: 952, 1991. The latter suggests:
Aldosterone/Renin > 920: Primary aldosteronism or maybe chronic renal failure
Nl. A/R, high aldosterone: Secondary aldosteronism
Nl. A/R, low aldosterone: Hyporeninemic hypoaldosteronism
Aldosterone/Renin <28:Addisonism
All others: Everybody else
The lucky patient with proven primary hyperaldosteronism has an adrenal cortical adenoma (find it with the CT scanner and/or differential concentrations of aldosterone in the adrenal veins). Surgery will be curative, and aldosterone-producing cancers are very rare.
Less common is "idiopathic adrenal hyperplasia" associated with primary aldosteronism. (In recent studies, 10-40% of patients with primary aldosteronism had no adenoma. The nodular cortices can simulate adenoma: Surg. 106: 1161, 1989.)
*Even less often, the patient has "glucocorticoid suppressible" primary hyperaldosteronism, which depends on the pituitary gland. See Am. J. Med. 72: 851, 1982; NEJM 306: 746, 1982 for more about this rare familial disorder. (Saline may suppress.)
Telling surgical disease (adenoma) from medical disease (hyperplasia) involves both scans and venous sampling in the radiology department. * In idiopathic hyperplasia (but not adenoma), serum aldosterone supposedly changes with posture, etc., etc.
TESTING FOR 21-HYDROXYLASE DEFICIENCY ("congenital adrenal hyperplasia"; Arch. Int. Med. 147: 847, 1987; NEJM 316: 1519, 1987)
The most obvious cases have preposterous elevations of 17-hydroxyprogesterone, and even mild cases will have an exaggerated 17-hydroxyprogesterone response to ACTH stimulation.
For the diagnosis of other congenital adrenal hyperplasia enzyme deficiencies, check with your endocrinologist.
ADRENAL MEDULLARY TUMORS (End. Metab. Clin. N.A. 17(2): 397, 1988; Mayo Clin. Proc. 65: 88, 1990; Cancer 62: 2451, 1988; Hosp. Pract. 24(1): 175, Jan. 1989.)
You will detect 98% or more of pheochromocytomas if you get a 24 hr urine specimen (use strong acid as the preservative for this assay) and order urinary catecholamines, metanephrines, and vanylmandelic acid (VMA).
Be sure to order a total creatinine on the sample to be sure it is complete.
Many drugs and possibly some foods interfere with the old colorimetric determination of various catecholamine metabolites. (Interferences with the newer HPLC techniques are much less of a problem.)
It is probably best for the patient to remain off all drugs and to avoid the foods listed above for the week prior to the test.
Several newer tests have been described to help make the diagnosis of pheochromocytoma easier.
Platelet catecholamines (taken up by platelets from the serum, of course) can be measured, though the value of this test is not yet established (NEJM 306: 890, 1983), and the test hasn't caught on.
Plasma normetanephrine levels are much higher in most patients with pheochromocytoma than in other hypertensives, and this assay does not appear to be affected by antihypertensive drugs. (Thus, this test might be suited for patients who cannot be taken off antihypertensive medication.) Metanephrine and * 4-hydroxy-3-methoxy mandelic acid are available as adjuncts (J. Clin. Path. 46: 280, 1993).
*Free norepinephrine and 3,4-dihydroxyphenylglycol (serum, urine) seem sensitive and specific (NEJM 319: 136, 1988). More on these Am. J. Med. 92: 147, 1992.
*Neuropeptide Y in serum may be available soon to screen for pheo, neuroblastoma, and ganglioneuroblastoma (Clin. Sci. 83: 205, 1992). Neuron-specific enolase in serum may also be useful to screen for, and follow, neuroblastoma (according to an obscure journal, but it makes sense: In Vivo 5: 245, 1991 abstract 91370744).
The clonidine suppression test is designed to distinguish high plasma catecholamine level due to pheochromocytoma from cases due to "stress", "white jacket syndrome", etc. It works on the principle that the drug fails to suppress catecholamine production by pheochromocytomas, while turning off the rest of the sympathetic nervous system.
A new protocol works on urine and sounds good: Am. J. Med. 84: 993, 1988. Review Arch. Int. Med. 152: 1193, 1992.
*A glucagon stimulation test, intended for patients with normal baseline plasma catecholamines, is not much used ("improved safety" through alpha-blockade: Arch. Int. Med. 149: 214, 1989).
Serum chromogranin A (stored and released with catecholamines) is an exotic tumor marker for pheochromocytoma (Medicine 70: 33, 1991).
An isotope scan (I-131-metaiodobenzylguanidine) is available, too (NEJM 305: 12, 1981; J. Urol. 134: 105, 1985). Angiography is hazardous, as it can result in massive release of catecholamines (uh oh!), but CT scanning is very good for finding "pheos".
*An article from the competition's surgeons on pitfalls of venous sampling to diagnose pheos: Am. Surg. 54: 632, 1988. Yet another protocol for pheochromocytoma suspects: Medicine 70: 46, 1991.
nCarcinoid tumors at many sites may be detected and monitored by measuring 5-hydroxyindoleacetic acid (5-HIAA), the principal metabolite of serotonin. (Review: Endo. Metab. Clin. N.A. 17(2).
ADRENAL INCIDENTALOMAS: The current rule seems to be that if you find a benign-looking adrenal nodule on CT scan, and it is >3 cm across, you must at least follow it with endocrine tests and repeat scans. ("And a chance to cut is a chance to cure": Am. Surg. 55: 516, 1989.)
(Clinical Pathology students: Learn the concepts, don't memorize the numbers.)
_