
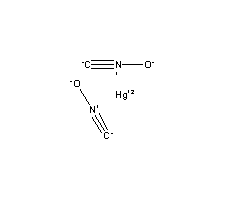
| Reagents | Apparatus |
| .5 mL Mercury, Hg | Graduated Cylinder |
| 47 mL 70% Nitric Acid, HNO3 | Filter Paper |
| 59 mL 96% Ethanol, C2H6O | Beaker/Round Bottom Flask |
| Funnel |
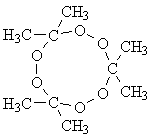
| Reagents | Apparatus |
| 300mL 3% Hydrogen Peroxide, H2O2 | Beaker |
| 100mL Acetone, C3H6O | Graduated Cylinder |
| 30-50mL Sulfuric Acid, H2SO4 | Filter Pape |
| Funnel | |
| Thermometer |
