|
"Understanding nature's mute but elegant language of living cells is the quest of modern molecular biology. From an alphabet of only four letters representing the chemical subunits of DNA, emerges a syntax of life processes whose most complex expression is man. The unraveling and use of this ‘alphabet’ to form new ‘words and phrases’ is a central focus of the field of molecular biology. The staggering volume of molecular data and its cryptic and subtle patterns have led to an absolute requirement for computerized databases and analysis tools. The challenge is in finding new approaches to deal with the volume and complexity of data, and in providing researchers with better access to analysis and computing tools in order to advance understanding of our genetic legacy and its role in health and disease."
From the National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
This project is a series of interrelated modules designed to introduce the student to modern biological techniques in the area of Bioinformatics. Bioinformatics is the application of computer technology to the management of biological information. The need for Bioinformatics has arisen from the recent explosion of publicly available genomic information, such as that resulting from the Human Genome Project. To address this, the National Center for Biotechnology Information (NCBI), at www.ncbi.nlm.nih.gov/, was established in 1988 as a national resource for molecular biology information. The NCBI creates public-access databases, develops software tools for analyzing genome data, and disseminates biomedical information - all for the better understanding of molecular processes affecting human health and disease. The NCBI is a virtual goldmine both in terms of available resources, and treasures yet to be discovered. Some of the many databases that the NCBI is responsible for include the GenBank DNA sequence database, containing over 9,103,000 nucleotide sequence records as of October 2000; the Molecular Modeling Database (MMDB) consisting of three-dimensional protein structures, as well as tools for their visualization and comparative analysis; and the Online Mendelian Inheritance in Man (OMIM) database, which is a catalog of human genes and genetic disorders.
The NCBI has developed a simple way to analyze and access all of this information. Entrez is a search and retrieval system that integrates the nucleotide sequences, protein sequences, macromolecular structures, whole genomes, and scientific literature (see below). The benefit of this system is that all of these databases are linked to each another, so that one may easily access a protein sequence, obtain literature on that protein, examine clinical disorders resulting from defect in the protein, and analyze the chromosomal location of the gene, all with the click of a button. It is a useful way to get started analyzing the incredible amount of information available through the NCBI.
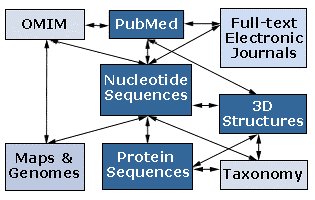
http://www.ncbi.nlm.nih.gov/Database/index.html
Feel free at any time to browse further into each site following your curiosity. If you ever get lost in the databases, you can always return to the original web address listed above.
By completing this project, you will be exposed to the tools and databases currently used by researchers in molecular biology, and you will gain a better understanding of gene analysis, and the relationship of amino acid sequence to protein structure and function. While no computer programming skills are necessary to complete the modules in this work, prior exposure to personal computers and the Internet will be assumed. The main program that you will need is an Internet browser, such as Netscape Navigator or Internet Explorer. You will also need a molecular modeling viewing program called RasMol, which may be downloaded (for free) or is also located on the Compaq Deskpro computers in the Computer and Information Services Lab (located right behind Hawes Hall). You should also have available an empty 3.5" computer disk with which to save a protein file (discussed later in this exercise).
MODULE 1. Taxonomy Revised 5/23/02
Begin by linking to the NCBI homepage at www.ncbi.nlm.nih.gov/ and familiarize yourself with the many selections on this main page. If you ever get lost, always return to this page as a starting point. Select the "Taxonomy" option (now called TaxBrowser) to be linked to the NCBI taxonomy homepage. The NCBI Taxonomy database contains the names of those organisms whose sequences have deposited. Sequence data are available for only a small amount of the approximately 2-10 million different species estimated to exist on earth. Select the option "How many organisms are represented at GenBank?" in the lower left frame.
- For the year 2001, how many different species are represented in the sequence databases?
Interestingly, the sequence data from even extinct organisms are also listed in the GenBank database. Select the option "Extinct organisms" at the lower left of the screen to see what organisms are listed in the databases. Nucleotide sequence information is available from such extinct organisms as Homo sapiens neanderthalensis, and Smilodon fatalis (saber-toothed cat). Select the option "Mammuthus primigenius" (woolly mammoth). The next page that is displayed gives you some very specific information concerning the lineage (ancestry) of this organism. Select the option "Proboscidea" in the lineage. You will be linked to a page containing information on this listing.
- Besides the wooly mammoth, what are some other organisms that belong in this classification order?
Amazingly, a number of nucleotide sequences have been deposited into the GenBank database from this organism. The deduced protein sequences of many of the DNA sequences are also listed.
- How many nucleotide sequences have been deposited into GenBank to date for Mammuthus primigenius?
Retrieve the nucleotide sequences for this organism by selecting "Submit Query". Select the accession number "D50842", which will display the complete coding sequence of the mitochondrial DNA coding for cytochrome b. You will have to go through several pages to get to this one. Page numbers are at the lower right.
- What role does the protein cytochrome b play in the body?
What follows as you scroll down the page is a complete reference report on the nucleotide and protein sequence of cytochrome b from an organism that lived a very long time ago. Amazing!
- Approximately how many years ago did the wooly mammoth become extinct?
- How many nucleotide base pairs (bp) does this DNA entry contain? (The answer is in the first line)
Scroll through the complete reference report on this DNA and deduced amino acid sequence. A lot of information may seem confusing, but it is all here to provide researchers with as much information as possible about this gene. Notice that the "Mammuthus primigenius [extinct] mitochondrial DNA, complete cds" contains the complete coding sequence (cds) and is from the mitochondrial DNA. Several "References" are also listed for this sequence. For example, under Reference 3, we see that this nucleotide sequence was deposited into the GenBank database in June 1, 1995. In addition, under Reference 1, we find a complete research article related to this sequence. Noro,M., Masuda,R., Dubrovo,I.A., Yoshida,M.C. and Kato,M. Molecular phylogenetic inference of the woolly mammoth Mammuthus primigenius, based on complete sequences of mitochondrial cytochrome b and 12S ribosomal RNA genes. J. Mol. Evol. 46 (3), 314-326 (1998). A direct Medline link "MEDLINE 98154407" to this article is also given, in case you wish to read the article abstract concerning this nucleotide sequence.
At the bottom of the screen, you will find the nucleotide sequence (all of the A, G, C, and T letters), as well as the deduced amino acid translation.
- What do we mean when we refer to a deduced protein sequence?
Notice that the protein identification number ""BAA25008.1"" is also listed next to the protein translation. Select this link to examine only the amino acid sequence.
- How many amino acids comprise this cytochrome b protein?
|
MODULE 2. OMIM, Online Mendelian Inheritance in Man
Select the OMIM Online Mendelian Inheritance in Man option at the NCBI homepage. This database is a catalog of human genes and genetic disorders, focusing on inherited, or heritable diseases. The power of this tool is that is will allow you to learn more about a clinical disease, and how that disease may be transmitted from parent to offspring.
Begin by searching OMIM for "hypercholesterolemia", one of the most common genetic disorders, and characterized by elevation of serum cholesterol bound to low density lipoprotein (LDL). Although many entries come up, select the one labeled "*143890 HYPERCHOLESTEROLEMIA, FAMILIAL".
You will be linked to a page containing a very detailed description and genetic analysis of Familial hypercholesterolemia, caused by mutations in the LDL receptor (LDLR) gene. Scroll through the pages or select the choices on the left most side of the screen (e.g. clinical features, cytogenetics, population genetics) and answer the following questions. An abbreviated version of this page is available at "MINI-MIM" and "Clinical synopsis" located at the lower left of the page.
- On what chromosome is the gene for the LDL receptor found?
- In addition to elevated serum cholesterol, what are some of the other maladies in those patients suffering from this disease?
- What is the relationship between the LDL receptor and hepatitis C virus?
- What is the frequency of familial hypercholesterolemia heterozygotes in the population?
- In terms of coronary artery disease, at what age would one expect to see heterozygotes showing symptoms of the disease? At what age for homozygotes?
|
MODULE 3. Protein Analysis
During the course of this module, you will learn more about a particular enzyme referred to as an apyrase, also referred to as ATP-diphosphohydrolase, or ATP-diphosphatase. As is the case with some enzymes, there are several names for this protein, depending upon who discovered the enzyme, who purified it and cloned it, who unknowingly purified the same enzyme but named it something else, etc...
Let’s learn more about this apyrase enzyme. Go to the NCBI homepage and enter the term "apyrase" in the blank and then select "GenBank" from the pulldown menu at the left. GenBank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences. There are approximately 10,336,000,000 bases in 9,103,000 sequence records as of October, 2000. Then click the "Go" button to begin the search (you are now searching the NCBI GenBank database for apyrases). A page of matches (131 matches as of 1-1-2001) will be returned to you indicating the number of documents that contain text matching "apyrase". Most of these entries are cDNA sequences from organisms such as soybeans, chickens, rats, potatoes, etc. While all of this information is useful to researchers in the apyrase field, we want to find an apyrase from a specific organism, like one called Aedes.
Go back one click to the original GenBank database search form of the NCBI and this time leave "apyrase" in the blank but add "Aedes" to this as well (no quotes). Now press the "Go" button. You should retrieve only 2 sequences this time. Click on the "L12389 Aedes aegypti apyrase mRNA, complete cds" to learn more about this enzyme. What follows is a lot of technical information concerning the Aedes aegypti apyrase.
- What is the common name of this organism? (You can link to the taxonomy site by selecting the organism).
- How many nucleotide base pairs (bp) does this mRNA entry contain? (The answer is in the first line)
Notice that the "Aedes aegypti apyrase mRNA, complete cds." contains the complete coding sequence (cds) and was deposited into this database in 1995. Notice also that there is a GenBank direct accession number for this mRNA (ACCESSION L12389).
There is more than one way to examine this apyrase mRNA. In particular, although the entire mRNA sequence is given, not all of the sequence consists of the coding region. That is, only a section of the total mRNA codes for a functional protein. For a clearer view of this, click on the "Display" button on the upper left portion of the page after selecting the "Graphics" icon from the pulldown menu at the top of the page. Scroll down a bit and notice that although the entire mRNA (represented by the nucleotides A, C, T, G) contains over 1700 base pairs, the pink coding sequence only runs from about nucleotide 20 to nucleotide 1710. The remainder of this sequence is called untranslated sequence (i.e. it is not translated as part of the protein).
This apyrase enzyme is also referred to as an ATP-diphosphohydrolase. This is because the enzyme functions as an extracellular nucleotidase, hydrolyzing nucleotides like ATP or ADP to AMP. There is an incredible amount of information concerning this enzyme available on this page for student scientists. Display the "Default View" again and select the "protein_id="AAC37218.1" option next to the amino acid translation to link to additional information concerning this apyrase protein. As you continue to scroll to the end of the page, you will notice the section that contains a lot of letters. This section is the translated protein product, showing the complete amino acid sequence of the apyrase protein. Note that only the single letter amino acid code is shown for space-saving reasons.
- How many amino acids (aa) is the apyrase protein composed of?
- What are the first five amino acids of the apyrase enzyme sequence? Write them out, don’t use the single letter abbreviations.
Scroll down the page to learn more about the features and amino acid sequence of this protein. For example, under the FEATURES section, you can see the sex distribution and tissue specificity of this enzyme in Aedes aegypti.
- In what type of tissue was the mRNA for this enzyme found in Aedes aegypti?
The apyrase gene from this organism was first cloned and described in a paper listed under reference #1. Notice that there is a Medline link to the scientific paper describing this apyrase. Link to MEDLINE "95148604". Here we can find the scientific abstract from the article concerning this protein.
- Please distinguish between hematophagy and hemostasis? In your own words, what do you think the function of this enzyme is and how is it related to hematophagy?
Fortunately, as is the case for some science journals, we can view for free the complete report describing this protein. Link to the "FREE full text article at www.pnas.org", and then "DOWNLOAD the reprint (PDF) version of this article". You will need Adobe Reader (download for free) in order to view the PDF file for this article. The following questions refer specifically to this article.
- What year was the term "apyrase" first reported in print, and by what author?
- What did the authors feed the Aedes aegypti organisms that they reared?
In Figure 4 of the article, evolutionarily related proteins are compared to the Aedes apyrase.
- The amino acid sequence of the Aedes apyrase is compared to the 5’ nucleotidase proteins from what two mammalian organisms?
|
MODULE 4. PubMed at the National Library of Medicine
MEDLINE is the National Library of Medicine's (NLM) bibliographic database. PubMed is the National Library of Medicine's search service that provides access to over 11 million citations in MEDLINE, with links to participating online journals. PubMed allows us to easily search for scientific and medical references. Begin by going to the PubMed homepage off of the NCBI website by clicking the "PubMed" option at the upper left portion of the page.
Once you are at the PubMed home page, you will want to do a specific search to easily eliminate undesirable scientific articles. If we want specific articles concerning say, fruit flies, the easiest way to approach this is to keep the default parameters "as is" and search for the scientific name of the "fruit fly" organism. To find the scientific name of the fruit fly, you must go to the Taxonomy homepage, and search for "fruit fly". There you will find the genus and species name of the common fruit fly.
- What is the scientific name (genus and species) for the fruit fly?
Once you know the genus name, you may type that into PubMed. Upwards of 34,000 articles come up when you search PubMed using the genus name (as of 1-1-01), most of which have an abstract available. That is a lot of articles to sift through on the fruit fly. Hmm, let’s get a little more specific. How about searching for a specific gene in fruit flies? A new gene (abbreviated Indy, for I’m Not Dead Yet) has recently been studied that has been shown to extend life span. Try searching for the genus name and the gene Indy as follows: "(genus) AND Indy". At least one paper will come up that has both the fruit fly genus name AND the word Indy somewhere in the title or abstract.
- What is the title of the article authored by Rogina B, Reenan RA, Nilsen SP, and Helfand SL?
Click on this article to be linked to the abstract to this recent Science journal article.
- When the single gene was mutated in the fruit fly, approximately how many times longer did the mutated fruit flies live, compared to the non-mutated flies?
- What did the authors speculate might be the mechanism of action for extending the lifespan of the flies?
Imagine the implications that this gene might have if a version of it was found in the human genome! What we would need to do next is to organize a search the human genome for homologs of this fruit fly gene.
|
MODULE 5. BLAST for Protein Homology
The databases at the NCBI will allow you to pose and answer questions like "What is the evolutionary relatedness (if any) of one protein to another?" In order to answer a question like this, one needs to be very familiar with the vocabulary of similarity searching (looking for similarities between one subject and a group of others). To become familiar with the premise, link to the "Education" homepage off of the main NCBI site (located at the lower left). On the far left of this "Bioinformatics" page, select the "Similarity searching" link. This page will provide you with enough information to understand the premise behind similarity searching and allow you to begin conducting searches.
- Define Homology.
Let us put these Basic Local Alignment Search Tools (abbreviated BLAST) to work to begin to look at the relatedness of proteins. For instance, let us look at that particular Indy protein produced by the fruitfly. Fortunately, the authors of manuscripts are required to include an accession number with which to locate any published gene or protein sequence. Since the accession number for this fruitfly protein was listed in the original Science article (accession number AAF49226), we can easily find this protein sequence to study it further, and ultimately find if any human homologs exist of this protein.
Begin by linking to the NCBI homepage and then select "Entrez" from the top menu. Once you have entered Entrez, select the "Protein" sequence database. At this point, search the protein database for the accession number above, "AAF49226". What you will find on the next screen is the protein product for that gene, AAF49226, CG3979 gene product [alt 1] [Drosophila melanogaster]. Click on the returned product. Congratulations, you’ve found the Indy protein!
Scrolling through this page you will find reference information for this protein. A prettier way to display this 572 amino acid protein is to display the sequence as a graphic, instead of just the single letter code. To do this, at the top left of the page, "Display" the "Graphics" format from the pulldown menu. The next page you see will show you the Indy protein represented by a green bar, with all of the single amino acids listed. Return to the previous page by selecting "Display" the "Default View" from the pulldown menu at the top of the page again.
If you scroll to the bottom of this original page, you will notice that a run of amino acids is shown. This is the primary amino acid sequence of the Indy protein from the fruit fly. To do any further analysis however, such as looking for related proteins in humans, we have to display this information in a form without spaces or numbers, in essence, without any extraneous information. To do this, at the top left of the page, "Display" the "FASTA" format from the pulldown menu. You will see a descriptive line (>gi|7293861|gb|AAF49226.1| CG3979 gene product [alt 1] [Drosophila melanogaster]) followed by the Indy amino acid sequence (without numbers, spaces, etc.). Highlight the entire amino acid sequence only, starting with the first M (Methionine) and ending with the last H (Histidine) by holding down the left mouse button and dragging the cursor. "Copy" this section using the browser’s copy tool listed under "Edit". Be sure that you have not copied anything but the amino acid sequence!!
Next you will use a program called BLAST (Basic Local Alignment Search Tool) to compare your copied fruit fly Indy protein sequence to all of the proteins reported from the human genome. To do this by hand would take quite some time, but by using the algorithms at the NCBI, it is amazingly fast! BLAST has revolutionized the way sequence information can be used.
After you have copied this protein sequence, return to the NCBI homepage by selecting the small window in the upper left corner of the screen entitle "NCBI". Next, select the "BLAST" button located at the top center of the page. You have now entered the BLAST search page. Select the "Advanced BLAST Search" under BLAST 2.1. and "paste" the complete Indy protein sequence into the largest window located in the center of the screen. Before submitting the request, several settings must be changed. The default "Program" used in this form is blastn (n=nucleotide). However, we’re working with a protein sequence here, not a nucleotide sequence, so we want to use blastp (p=protein). Pull down the "blastn" option and select "blastp".
Next, ensure that the Database button is showing "nr", for a non-redundant database. Scroll to the middle of the page (Advanced options for the BLAST server) where it says "or choose an organism from the list to limit your BLAST search:". Select "Homo sapiens" from the box, the scientific name for us humans. Scroll to the very bottom of the form and check the box indicating that you want the data to be returned "In HTML format". Now press the "Search" button. You will then be taken to the search page where you will notified that "Your request has been successfully submitted and put into the Blast Queue". Your "query" is the Fruit Fly Indy protein sequence, consisting of 572 "letters" or amino acids. You will also be informed of the estimated time that it will take to complete your search. Click "Format Results" and let the supercomputer at NCBI blast away at. Note that a separate browser will open up, and display your results. Be Patient.
When you get the results back they will be in a new website window in the form of a long data page. Just below some header information and a pretty colored chart is the table of related amino acid sequences of humans. Matches are scored based on the length of the amino acid match and the number of amino acid mismatches (variations) in that region. Low E-values (e.g. 0 or e-68 are VERY low numbers) indicating that the match is not likely to be due to chance alone. An E-value closer to or higher than 1 indicates a random match. Hence, although there are 17 "Hits" in the database, only some of these are truly related sequences indicated by their very low E-values.
Continue to scroll down the page, past the colored chart showing the database "Hits". These colored bars indicate the relative similarity of your query sequence to these human sequences. You will find a listing of all of the "sequences producing significant alignments". To look at how your query "Indy" sequence compares to these other human proteins, begin by clicking on the "Score" located to the right of each descriptive protein.
Choose the topmost sequence score "ref|NP_003975.1| solute carrier family 13 (sodium-dependent..." by clicking on the Score "319". You will see the actual matching regions of the renal sodium/dicarboxylate cotransporter (Sbjct sequence) with the Indy sequence (Query sequence). The center line between the subject and query is the consensus (match). There are also links to the respective Genbank entries. The number of identical amino acids between the Human sodium/dicarboxylate cotransporter and this Indy protein are given.
- What is a sodium/dicarboxylate cotransporter? Selecting the PubMed link to this protein may help you.
- What is the percent identity between your query sequence and the first sodium/dicarboxylate cotransporter protein listed?
As you continue to scroll through the matches of the Indy protein to the other human proteins, you can see their percent identity at the amino acid level.
At some places in the comparison between the two sequences, there is no amino acid letter in the middle line.
- What does the absence of an empty letter in the consensus line mean?
At some places in the comparison between the two sequences, there is a "plus" sign in the middle line, in place of a consensus letter.
- What does the plus sign indicate in the consensus line?
So it appears as if the human sodium/dicarboxylate cotransporter is related to some extent to the protein product of the Indy gene.
|
MODULE 6. Genes and Disease
With the rapid strides being made by the Human Genome Project, the location of many more genes involved in human disease are being identified. To examine this further, link to the NCBI website and select the "Genes and disease" icon under the rightmost column labeled "Hot Spots". You have now entered the section of NCBI that looks at the relationships between the 24 human chromosomes (22 autosomes, 2 sex chromosomes) and genetic diseases. On the lefthand side of the page, are some of the many categories that genetic diseases fall into. For instance, select the "Transporters" option at the lower right to learn about genetic diseases involving membrane carrier proteins and channels. Next select the "Cystic fibrosis" option at the upper left hand part of the page to learn more about this most common fatal genetic disease in the United States.
- What chromosome has the gene for CFTR (cystic fibrosis transmembrane conductance regulator) been localized to?
Notice that a number of other databases (like PubMed, OMIM) are linked to this page. Each one of these links would tell you more about this genetic disease cystic fibrosis. Return to the main "Genes and Disease" webpage.
At the top of the page, select the individual chromosome that the human CFTR gene is localized to. What is shown next is a set of chromosomes with genes identified as playing roles in human diseases. Clicking on each respective disease brings up information regarding that condition. For example, PENDRED SYNDROME is an inherited disorder that accounts for as much as 10% of hereditary deafness, and the gene for this disorder is also localized to this chromosome.
- What is Williams Syndrome?
- In Williams syndrome individuals, both the gene for the protein __________ and an enzyme called __________ are deleted. What is thought to be the consequence for the individual through the loss of the former protein? What is thought to be the consequence for the individual through the loss of the later enzyme?
Many other genetic diseases have been localized to human chromosomes as well. Feel free to explore other chromosomes and the genetic disorders that have been localized to them.
|
MODULE 7. Molecular Structure and Visualization
The next part of this assignment is designed to introduce you to the basic concepts of protein structure by allowing you to look at and manipulate a 3-dimensional model of a protein. For instance, let us look at a structural protein found in a particular type of virus called Ebola virus. The Ebola virus is a member of a family of RNA viruses known as filoviruses. Ebola virus was first discovered in 1976 and was named for a river in Zaire, Africa, where it was detected. Specifically, let us look at a particular protein of the Ebola virus called GP2. The 125 kD glycoprotein (GP) is an integral membrane protein and forms the surface projections of the virion. It is reasonable to assume that the glycoprotein is the mediator of virus entry into the cell. Functional sites for receptor recognition and binding and perhaps for fusion should be located on this protein. We will be able to explore the molecular structure of the Ebola virus glycoprotein. Much more information concerning the Ebola virus can be found online, through the Centers for Disease Control (CDC) website at
http://www.cdc.gov/ncidod/dvrd/spb/mnpages/dispages/ebola.htm
In this exercise you will examine the Ebola GP protein using a program called RasMol which allows you to not only visualize a model of this complex protein, but also to rotate, and highlight various features of the protein. The models are generated by the program RasMol but are based on atomic coordinates stored in the Protein Data Bank at Brookhaven National Laboratories http://www.rcsb.org/pdb/. The Protein Databank is an international repository for the processing and distribution of 3-D macromolecular structure data primarily determined experimentally by X-ray crystallography and NMR. This means that over 11,600 3-D macromolecules like proteins, peptides, viruses, nucleic acids, and carbohydrates are stored right there. You can search for those stored compounds (like poliovirus, collagen, or DNA) using the "SearchLite" icon located on the right hand side, or if you know the specific accession number for your molecule of interest, you can simply type that in the space indicated. NCBI's structure database is called MMDB (Molecular Modelling DataBase), and it is a subset of three-dimensional structures obtained from the Brookhaven Protein DataBank. It is the NCBI’s Molecular Modelling DataBase that we will use to search for 3-D protein structures.
To begin the analysis, at the NCBI homepage select the "Structure" option. Next, type in the word "Ebola" where it says "Search Entrez Structure for" and click "Go". The next page shows you the information concerning the available proteins to be examined from the Ebola virus. Select the "1EBO Crystal Structure Of The Ebola Virus Membrane-Fusion Subunit, Gp2, From The Envelope Glycoprotein Ectodomain". You will now be linked to the MMDB structure summary page.
- When was the structure of this protein deposited in the Protein Data Bank (PDB)?
Notice that there is a Medline link to read more about the crystal structure of this protein. Link to the article by selecting "PubMed" and then select the article to pull up the abstract and technical information concerning the crystallization of this protein.
At this point, we want to view the structure of the Ebola protein. In order to do this, click on the option "View/Save Structure" located on the left-hand side of the screen, as well as the Option "Save File". It is important that you rename the file "1EBO.pdb" and save it to the empty 3.5" disk that you should have with you.
At this point, assuming you have successfully saved the Ebola protein file, you can shut down the Internet browser. You will not need it for the remainder of the exercise. Next open the RasMol program already installed on the Compaq Deskpro computer in CIS. On the desktop screen, go to the far-left corner and click the "Start" button. Highlight the "Programs" line and then scroll over to the "Biology-Chemistry" line. At the bottom of the 5 or 6 programs listed under "Biology-Chemistry", select "Rasmol". What you will see next is an empty black display screen that opens (this is the RasMol version 2.6 display screen), as well as a small white box (the RasMol Command Line). At this point, "Maximize" the RasMol display screen, or completely open it to fill the entire screen. This is the screen with which you will view the 3-D protein structure in and in which nearly all steps will be performed. The RasMol Command Line will be located at the bottom of the screen.
Now go to the pull down menu at the top and select "File" and then "Open" the "1EBO.pdb" file that is located on the 3.5" disk that you saved it on (hopefully). After a few moments, a file will be opened that looks like a jumble of colored sticks on the RasMol screen. Congratulations, you’ve succeeded in opening and are now looking at the atomic structure of the Ebola glycoprotein!!
When the Ebola structure is loaded, next view it in all of the available RasMol display modes starting with the default display, "wireframe". To change the view, pull down "Display" and select "backbone", "sticks", "spacefill", "ball & sticks", "ribbons", "strands", and "cartoons". These various displays highlight different aspects (like sidegroups, helices, sheets) of the proteins.
Rotate the structure by moving the mouse while holding the LEFT mouse button down.
Zoom in or out on the structure by moving the mouse up or down while pressing the Shift" key and holding the LEFT mouse button down.
Change the "Display" to "backbone" to highlight only those bonds in the backbone of the molecule (side groups are not visible).
- How many subunits (Chains) is this Ebola protein composed of? You may need to rotate the enzyme to see how many polypeptide "ends" or termini exist.
Change the "Display" to "spacefill" to highlight those bonds in the backbone of the molecule as well as the amino acid side chains. Notice the different colors of the amino acid side chains (e.g. some are yellow)
The default "colour" used by RasMol is "CPK" in which the protein is colored by atom (carbon=grey, oxygen=red, nitrogen=blue, sulfur=yellow, and hydrogen=white)
Using the "Display" set the "cartoons" mode, and change the "Colours" from the default which is "CPK" to "structure" in which alpha helices are pink, beta sheets are yellow, and beta turns are blue.
- How many regions of alpha helices are there in the Ebola glycoprotein?
- How many regions of beta sheets are there in the Ebola glycoprotein?
Visualize the 599 hydrogen bonds that help to stabilize the secondary and tertiary structure of the Ebola fusion protein by selecting the RasMol Command Line. In the Command Line window RasMol> type "hbonds" (no quotes). Return to the RasMol display screen by selecting it at the bottom of the screen or clicking on the black window. The hydrogen bonds are best seen in "Display" set to "ribbons", "Colour" set to "CPK". A single hydrogen bond in this view appears as a red/blue dotted line. Rotate the image to examine the locations of these bonds.
- Do hydrogen bonds appear mostly within single alpha helices or between adjacent alpha helices?
- Do hydrogen bonds appear mostly within single beta sheets or between adjacent beta sheets?
Turn off the hydrogen bonds by typing "hbond off". Return to the "wireframe" display. Set the "Colour" to CPK. How about looking at some specific amino acids in this Ebola glycoprotein? To do this, you can go to the RasMol Command Line and "select" any amino acid you would like by using the three letter amino acid abbreviation. For example, type "select pro", which tells the program to now focus all subsequent commands on the pro residues alone. Under "Display" chose "spacefill". What you now see are enlarged proline residues in the protein.
To return to the previous view, choose the "Edit" menu and click on the "Select All". This now tells the program to focus on all amino acids again. Now set the "Display" to "wireframe" to return to the same old view.
- Using the protocol that was just described, identify how many cysteine residues are in this protein. Remember that you must use the three letter amino acid code when doing this.
Return to the previous view by choosing the "Edit" menu and click on the "Select All". This now tells the program to focus on all of the amino acids again. Set the "Display" to "backbone". Next select the cysteine residues again and then display them as "ball and stick" to better visualize them. Notice that the yellow ball indicates sulfur atoms.
The closeness of these cysteine residues suggests that they may be involved in disulfide bond formation. Visualize the disulfide bonds by selecting the command line window and typing "ssbond". You may have to look very closely and rotate the molecule in order to see if any disulfide bonds are present.
- How many disulfide bonds are present in this molecule?
The crystal structure of the Ebola glycoprotein was determined with several hetero atoms included. Highlight these additional atoms by typing "select hetero" (no quotes) in the RasMol Command Line. You have told the program to now focus all subsequent commands on these atoms alone. Under "Display" chose "spacefill". Several differently colored atoms are now shown. Clicking on each of these atoms individually, and then examining the RasMol Command Line, will help you identify each ion.
- What atom is represented by the green colored ball?
|
Well, that’s about it. You have successfully mastered some of the state-of-the-art tools used by most molecular biology researchers today. |

