AGENTES CANCERÍGENOS Y EVOLUCIÓN DE UN TUMOR
Puede hablarse de dos tipos de agentes responsables de la primera alteración celular:
El humo del tabaco pertenecería al primer grupo, ya que contiene numerosas sustancias que son iniciadores o promotores tumorales: benzopirenos, nicotina, naftilaminas, fenoles... mientras que las hormonas, como los estrógenos, lo harían al segundo.
Es la segunda fase, promoción, durante la cual el agente promotor estimula el crecimiento de las escasas células iniciadas que con una sola mutación tenían ligeramente alterado su crecimiento. Este aumento de células con una mutación favorece la posibilidad de que alguna de ellas acumule una nueva mutación que la haga crecer aún más deprisa, ya que la división celular aumenta el riesgo de adquirir mutaciones.
La incidencia de tumores y su velocidad de progresión hacia la malignidad dependen de la frecuencia de mutación. Así, una velocidad de mutación incrementada surge por la presencia en el medio de agentes mutagénicos, por defectos en la capacidad de división celular, en una capacidad defectuosa de reparación del DNA dańado etc. La reducida probabilidad de mutaciones espontáneas hace que la duración de esta fase en que el tumor no es aún visible sea muy larga, puesto que se necesitan millones de células con una mutación para que alguna desarrolle un segundo cambio genético. Conocemos muchas mutaciones que explican el aumento en la proliferación celular, pero aún muy poco sobre las mutaciones responsables de la gran capacidad invasiva que presentan las células tumorales.
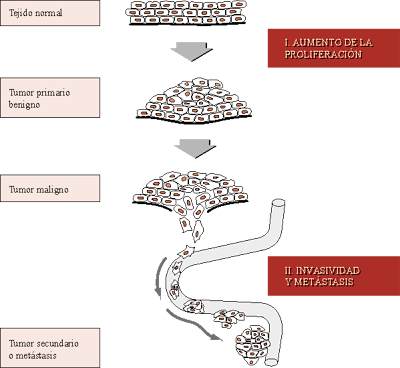
La tercera fase es la progresión tumoral o adquisición de nuevas (tercera, cuarta...) alteraciones genéticas que provocan un aumento de la malignidad, con adquisición de capacidad invasiva y metastásica.
La progresiva acumulación de mutaciones, probablemente en una secuencia específica para cada tipo celular, origina primero una hiperplasia o crecimiento desordenado, y luego invasividad celular, vascularización o angiogénesis y, finalmente, metástasis.
Algunos agentes cancerígenos transforman una célula madre sin producir un tumor, pero este cambio es transmitido a la descendencia hasta que una "seńal" provoque la proliferación irregular y la cancerización. Esta seńal desencadenante puede ser muy diversa: desde una infección, pasando por un cambio hormonal o un traumatismo. Algunos autores afirman, incluso, que es posible que se de un proceso de cancerización sin la existencia de mutación, ya que la consideran como una tendencia natural de toda célula, expresada al cesar los controles de ejercer su función.
DEFENSAS DEL ORGANISMO CONTRA EL CÁNCER
Existen varios mecanismos de defensa contra el cáncer desde el propio organismo:
EN QUÉ SE DIFERENCIAN LAS CÉLULAS CANCEROSAS DE LAS NORMALES
Ya se ha dicho que una célula cancerosa se caracteriza por su independencia con respecto a las seńales reguladoras del medio y su división incontrolada, pero también posee otras muchas características que le sirven como estrategias para su supervivencia. Una célula cancerosa fabrica sustancias que no le corresponden y tiene una extraordinaria movilidad. A diferencia del resto de células, puede resistir las condiciones que se dan en la circulación sanguínea, lo cual posibilita su diseminación eficiente por todo el organismo. Además, construye defensas contra las seńales reguladoras que recibe, la respuesta inmune desarrollada en su contra y los medicamentos que pretenden atacarla. Pero ciertamente la invasividad es, la característica esencial del cáncer.
Las estrategias de las células cancerosas pueden resumirse del modo que sigue:
1-acentuación progresiva de la malignidad con cada división
2-proceso de selección dentro de un tumor, así sobreviven las células más adaptadas, que son aquellas más mutadas y, por tanto, más maligna
3-los tumores producen enzimas capaces de destruir las membranas que rodean los tejidos para así invadirlos
4-incremento de su motilidad
5-capacidad de atravesar el endotelio vascular y así entrar en la circulación sanguínea
6-resistencia a las variaciones de la tensión de oxígeno entre los tejidos periféricos
7-capacidad de agregación plaquetaria por producción de trombina para producir coágulos sanguíneos que detengan a las células cancerosas en los capilares, para luego pasar al tejido y colonizarlo
8-resistencia a la respuesta inmune mediante la expresión del lingando Fas en su superficie, el cual provoca la apoptosis de las células del sistema inmunitario
9-fabricación de factores de crecimiento por vía autocrina
10-expresión del receptor de IGF-1 para captarlo de la circulación (aunque la célula no perteneciera originariamente a un tejido capaz de hacerlo)
11-modificación de las células circundantes vía estimulación paracrina para que fabriquen factores de crecimiento, típicamente macrófagos (prostaglandina E2) y fibroblastos
12-emiten una seńal molecular que estimula la neoangiogénesis, así se procuran oxígeno y nutrientes (también pueden hacer que sean los fibroblastos quienes emitan la seńal)
13-resistencia a los tóxicos mediante hidrólisis o exocitosis
14-algunas entran en latencia (incluso durante 20 ańos) y son activadas por un cambio hormonal, una infección, una intervención quirúrgica, una traumatismo... en este estado son insensibles a la quimioterapia
Hoy se cree que el proceso de aparición de células invasivas comienza en una fase relativamente temprana del crecimiento del tumor primario. Es decir, que la metástasis requiere tumorgenicidad e invasividad, procesos que se inician en este orden pero que se solapan parcialmente, de modo que mutaciones que proporcionan a las células tumorales la capacidad de invadir y formar posteriormente metástasis se comienzan a acumular ya durante la fase de crecimiento del tumor primario.
El crecimiento de un tumor, tanto primario como secundario, mayor de 5-10 mm de diámetro requiere su vascularización. Sin la cercanía de vasos sanguíneos, las células tumorales no sólo no pueden diseminarse, sino que mueren por deficiencia de nutrientes y oxígeno, y falta de eliminación de anhídrido carbónico, ácido láctico y otras sustancias de desecho debido a que los intercambios por simple difusión no alcanzan a las células internas del tumor. La neoangiogénesis, o formación de nuevos vasos sanguíneos a partir de otros preexistentes, es fundamental tanto al comienzo como al final del proceso de carcinogénesis. Al principio, para el aumento del tamańo del tumor primario y para el acceso al torrente circulatorio de las células tumorales, lo que permitirá su diseminación. Y al final, cuando alguna célula ha sobrevivido a la extravasación y coloniza tejidos distantes, para poder reiniciar el crecimiento de nuevos tumores secundarios.
Una vez en la cercanía de los vasos, las células tumorales deben atravesar sus paredes para acceder a la circulación sanguínea o linfática, que, en todo caso, están conectadas. Durante este proceso, las células deben soportar deformaciones que dependerán de la rigidez de las paredes y la disparidad entre su propio diámetro y el del vaso. Ya en el torrente sanguíneo (generalmente los sarcomas) o linfático (los carcinomas), las células tumorales deben sobrevivir al probable ataque del sistema inmune, adherirse a la pared de los vasos en una región distante y extravasarse. El establecimiento de metástasis por las células tumorales que han conseguido alcanzar la circulación sanguínea o linfática es un proceso afortunadamente muy poco eficiente. Se ha estimado que menos de un 0,05% de ellas lo consiguen. El riesgo de metastásis depende, naturalmente, del número de células del tumor primario que alcanzan el torrente circulatorio. Se ha calculado que un tumor primario de 1 cm de diámetro puede dar lugar al paso de millones de células a la sangre cada día. Para extravasarse, en primer lugar, las células deben quedar paradas en los microcapilares. En esta fase, las fuerzas mecánicas pueden ser más importantes que la formación de auténticas uniones adhesivas. Cabe esperar que grupos de células tumorales sean atrapadas con mayor facilidad en los microcapilares que las células individuales. Tras evadir la respuesta inmunológica y extravasarse, las células tumorales inician una segunda etapa de proliferación descontrolada y angiogénesis para dar lugar a un tumor secundario o metástasis.
El hecho de que el sistema inmune no sea capaz de detectar los sutiles cambios producidos en las proteínas de superficie de las células tumorales supone una ventaja más para las células cancerosas, las cuales, además, enmascaran sus antígenos. Este aspecto se rebela al comprovar que la mayoría de pacientes tiene unos niveles de sistema inmune normales al principio de la enfermedad. Puede afirmarse que es la progresión del cáncer el factor debilitante del sistema inmune y no la debilidad de éste el "causante" de la cancerización.
EL CICLO CELULAR
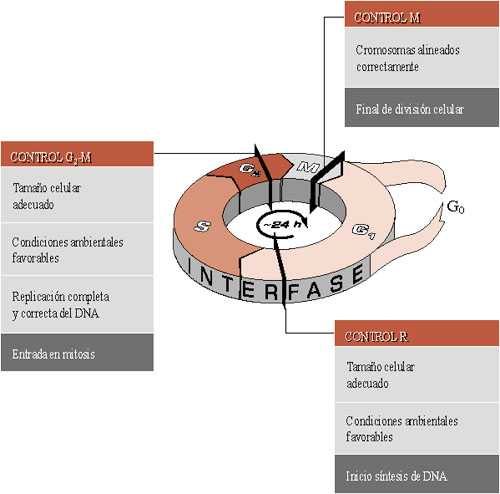
La proliferación celular puede estar regulada directamente a través de los puntos de control dentro del ciclo celular o por la muerte celular programada. La serie de procesos (a veces también el período que abarca) por los que una célula da lugar a dos células hijas se denomina ciclo celular. Consta de cuatro fases: G1, S, G2 y M . En su conjunto, y aunque hay variaciones, el ciclo completo dura unas 24 horas.
La fase G1 (del inglés "Gap" o intervalo) es el período de 6-12 h que sigue a una división celular y es previo a la síntesis o replicación del DNA. Durante este tiempo, la célula dobla su tamańo y masa debido a la continua síntesis de todos sus componentes como resultado de la expresión de los genes que codifican las proteínas responsables de su fenotipo particular. Hay células que pueden parar su progresión hacia la división en este estadío y permanecer durante días, meses o ańos en estado de reposo sin aumento de masa, en lo que se ha denominado fase G0. En la fase G1 existe un punto de control llamado el punto de restricción R en el que la célula comprueba que ha generado la masa necesaria para seguir adelante y comenzar la síntesis de DNA y, también, que las condiciones ambientales son favorables: presencia de nutrientes, sales y temperatura adecuadas; y de factores que induzcan crecimiento. Es el punto de control más importante.
La fase S (de "Síntesis" del DNA) corresponde al tiempo (6-8 h) durante el cual se replica el DNA. Cada cromosoma pasa a tener dos cromátidas. El período comprendido entre la finalización de la replicación del DNA y el inicio de la división es la fase G2 (3-4 h). Durante ella, las células se preparan para la escisión en dos células hijas. En esta fase existe un segundo punto de control G2-M, en el que la célula debe comprobar dos condiciones antes de dividirse: que ha duplicado la masa de modo que puede dar lugar a dos células hijas, y que ha completado la replicación del DNA, y sólo lo ha hecho una vez. Finalmente, las células entran en la fase de mitosis (1 h) propiamente dicha. En este momento existe otro punto de control M, que sólo permite seguir adelante si todos los cromosomas están alineados sobre el huso. Si esto es así, se da finalmente la citocinesis, que separará a la célula original en dos nuevas células.
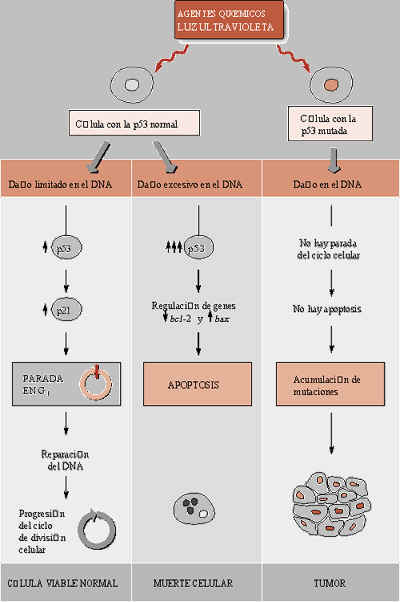
Existen controles negativos de la proliferación, potencialmente muy importantes para la prevención del cáncer, que se activan para parar el ciclo cuando se ha dańado la integridad del genoma y evitar así la aparición de células que puedan convertirse fácilmente en cancerosas. Muchos carcinógenos químicos y radiaciones actúan dańando el DNA o el sistema de microtúbulos necesario para la mitosis. Sin embargo, también causas internas pueden ocasionar alteraciones en el DNA, como los procesos de reordenamientos genéticos que tienen lugar durante el desarrollo, o los procesos de apoptosis o muerte celular programada, cuando las células tienen un DNA parcialmente degradado por acción de nucleasas que producen cortes en la molécula, o cuando las células están envejeciendo y se acortan los extremos de sus cromosomas (telómeros) provocando su inestabilidad. Estos controles, que coinciden con los puntos de control R y G2-M del ciclo, son críticos para evitar la inestabilidad genética que pueda llevar a la aparición de cáncer. De acuerdo con ello, en las células cancerosas están relajados o incluso faltan totalmente.
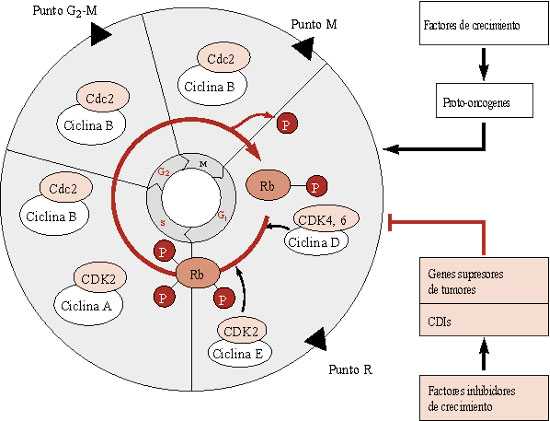
El sistema de control del ciclo celular se basa en dos familias clave de proteínas:
Las ciclinas reciben su nombre por la síntesis y degradación cíclica que sufren a lo largo del ciclo celular. Existen dos tipos de estas proteínas:
En mamíferos, además, hay al menos dos proteínas CDk diferentes, una para cada punto de control.
Así el centro del sistema de regulación del ciclo celular sería la asociación progresiva de Cdk con moléculas de ciclina para desencadenar los diferentes procesos subordinados del ciclo.
La síntesis de ciclina lleva a su acumulación progresiva, para formar junto a Cdk el complejo conocido como MPF (factor promotor de la fase M), el cual inicialmente permanece inactivo y posteriormente se activa por medio de fosforilación mediada por otras enzimas. La activación de MPF es casi explosiva, probablemente debido a un mecanismo de retroalimentación positiva del mismo MPF sobre las enzimas que lo fosforilan. Cuando su nivel es máximo, desencadena una serie de sucesos que conducen a la célula hacia el siguiente paso en el ciclo celular. Su inactivación también es muy acelerada devido a una rápida degradación de las ciclinas.
IMPLICACIÓN DEL ESTRÉS OXIDATIVO
La producción de especies reactivas del oxígeno durante las reacciones químicas celulares representa un grave problema para la célula, ya que éstas tienden a reaccionar con muchas estructuras, pudiendo así dańar a la célula. En este sentido, la célula posee su propia defensa: las enzimas antioxidantes, como la peroxidasa (que disgrega el peróxido de hidrógeno, un elemento altamente reactivo). El problema surge cuando las enzimas están en insuficiente cantidad en relación con los radicales libres.
El estrés oxidativo activa el gen llamado Bcl-2, cuya función es organizar resistencias a los dańos del DNA y promover la supervivencia celular, más que la apoptosis. Esto puede suponer un arma más para las células tumorales en las que el estrés oxidativo haya favorecido una elevada tasa de mutación.
Los genes llamados JNKS y Sek-1 son activados por estrés, anoxia, cambios osmóticos, procesos inflamatorios... Su función es la de activar los genes de defensa de la célula.
En bacterias existe el llamado sistema S.O.S, que reacciona a los dańos en el DNA para defender a estos organismos unicelulares. La proteína Lex-A se encuentra de manera normal reprimiendo una gran cantidad de genes; cuando se dańa el DNA, la célula pasa por un periodo de escasez nutricional ect, la proteína Rec-A destruye a Lex-A y además incrementa las tasas de mutación y autoriza (al mutarse los genes de reparación) la proliferación. Entre los genes activados se encuentran algunos que protegen frente a los choques térmicos y tóxicos. Se ha encontrado similitud entre la proteína Rec-A y la humana llamada Rad-51. Las bacterias también poseen el equivalente para el gen Bcl-2 humano.
LA ENZIMA TELOMERASA Y EL CÁNCER
Los telómeros son secuencias situadas en los extremos de los cromosomas eucarióticos lineales, a los que ayudan a estabilizarse.
Estas secuencias que son similares entre sí en organismos tan diferentes como los protozoos, los hongos, las plantas y los mamíferos, consisten generalmente en muchas copias en tándem de una secuencia oligonucleotídica corta, normalmente de la forma Tx Gy en una hebra y Cy Ax en la hebra complementaria, en donde x e y se encuentran típicamente en el intervalo de 1 a 4. Los telómeros mejor caracterizados son los de los eucariotas más simples. Los de levadura terminan con aproximadamente 100 pares de bases de secuencias repetidas de forma imprecisa y que se presentan como
(5f)(Tx Gy)n // (3f)( Ax Cy)n
En el momento del nacimiento, los telómeros de las células somáticas están formados por aproximadamente 15,000 pares de bases, que se van perdiendo en cada división celular a un ritmo de 25 a 200 pares de bases de la porción terminal del telómero. Cuando este fenómeno se repite 100 veces, la célula cesa de dividirse y muere.
Los extremos de una molécula lineal de DNA no pueden ser replicados por la maquinaria de replicación celular (y en ello puede radicar la causa de la circularidad de las moléculas de DNA bacteriano). Las secuencias repetidas de los telómeros se ańaden a los extremos de los cromosomas por acción de enzimas específicos, uno de los cuales es la telomerasa. No se conoce cuál es el factor que controla el número de repeticiones presente en un telómero.
El DNA que forma los telómeros está empaquetado en una forma de cromatina especialmente inaccesible, y se cree que ese tipo de empaquetamiento es el responsable de mantener, por ejemplo, al gen ADE2 translocado inactivo, proceso denominado silenciamiento por efecto de posición (la actividad de un gen es dependiente de su posición en el genoma)
La estructura de los telómeros plantea un problema biológico especial. La replicación del DNA requiere un cebador, pero en una molécula de DNA lineal es imposible sintetizar un cebador de RNA que empiece en el último nucleótido y reemplazarlo mediante los mecanismos normales. Sin un mecanismo especial para replicar los extremos, los cromosomas se acortarían algo en cada generación celular. El problema se soluciona con un enzima denominado telomerasa , que incorpora telómeros a los extremos cromosómicos. Aunque no sea sorprendente la existencia de este enzima, su modo de actuar no tiene precedentes. La telomerasa, lo mismo que otros enzimas descritos, contiene tanto RNA como proteína. El componente RNA tiene una longitud de unos 150 nucleótidos y contiene alrededor de 1,5 copias de la repetición telomérica Cy Ax adecuada. Esta parte del RNA actúa de molde para la síntesis del la hebra Tx Gy del telómero. En efecto, la telomerasa es una transcriptasa inversa que sólo sintetiza un segmento de DNA complementario a un molde de RNA interno.
La síntesis del telómero requiere un cebador Tx Gy corto y discurre en la dirección usual 5'--3'. Una vez sintetizada una copia de la repetición, el enzima se ha de reposicionar para reemprender la extensión del telómero. No se conocen aún los mecanismos mediante los que se termina este proceso cuando se ha sintetizado un telómero de longitud suficiente y de qué modo se sintetiza la hebra complementaria. Un rasgo de las secuencias ricas en guanilato, sin embargo, es que son capaces de replegarse sobre sí mismas formando pares de bases G-G diferentes del tipo Watson-Crick. Es posible que la hebra se pliegue sobre sí misma de esta forma cerca del extremo proporcionando un cebador para la síntesis de la hebra complementaria. Este proceso podría ser como sigue.
Esta estructura resultado del apareamiento no estándar G-G podría tener profundas implicaciones biológicas. La serie de Gs presente en los telómeros que genera este apareamiento podría mantener los cromosomas juntos en la meiosis.
Otro modelo, también basado en Tetrahymena, postula que la cadena retrasada se completa con la acción de la DNA polimerasa.
asa a, que porta la actividad primasa en una de sus subunidades.
Dado que el proceso de recorte y recuperación de las secuencias del telómero está equilibrado sólo aproximadamente, cada extremo de cromosoma contiene un número variable de repeticiones en tándem, que, en general, son de varios cientos de pares de bases de longitud.
Además de la replicación del telómero, la enzima telomerasa está involucrada en la reparación cromosómica, tal como quedó demostrado al comprobar que los cromosomas sometidos a repetidos ciclos de cortes y fusión a veces espontáneamente curan como si hubieran regenerado el telómero. Esta habilidad de reparar depende del tipo de tejido en el que se localiza el cromosoma alterado, y el fenómeno ha sido descrito en diversas especies. Incluso se ha reportado casos en humanos como el de los portadores de talasemia que tenían el cromosoma 16 con terminaciones truncadas, a la que se les adicionó terminación TTAGGG de novo. Se presume que un cromosoma roto fue reparado por la telomerasa y estabilizado a través de varias meiosis y mitosis.
L a pérdida de la actividad telomerasa en los protozoos (como Tetrahymena) da lugar a un acortamiento gradual de los telómeros con cada división celular, lo que lleva a la muerte de la línea celular. Existen dos teorías, que se complementan, para explicar el freno o "stop" de la proliferación celular coaccionado por el acortamiento gradual del telómero. Un modelo postula que el acortamiento límite del telómero es interpretado por la célula como un dańo del DNA, seńal para inducir la expresión del p53 y detener el ciclo celular. Otro modelo postula cambios en la expresión de genes reguladores localizados en áreas subteloméricas. Estos dos modelos no son mutuamente excluyentes y por el contrario ambos pueden contribuir a la iniciación del proceso de senescencia.
En las células germinales se mantiene la longitud de los telómeros pero no así en las células somáticas. Existe una relación lineal inversa entre longitud de los telómeros en fibroblastos cultivados y la edad del individuo del que se tomaron los fibroblastos: los telómeros en las células somáticas humanas se acortan gradualmente a medida que el individuo envejece. Una consecuencia es que las células germinales contienen actividad telomerásica pero las células somáticas carecen de ella.
Hace poco tiempo que se tienen evidencias, que provienen de estudios de cultivo de tejidos, que sugieren que la activación de la telomerasa podría permitir el continuado crecimiento de células inmortalizadas.
Los caminos para la activación de la telomerasa en tumores no son los mismos que en cultivo de tejidos. En cultivos primarios de fibroblastos humanos, la telomerasa no es activa, y como estas células se dividen, los telómeros se van acortando y las células entran en senescencia. La selección de las células inmortales se consigue por la expresión de un oncogen viral en las células pre-senescentes. Esta activación de la telomerasa se podría dar por mutación o reordenamientos que ocurren en estas líneas de células inestables. Los datos sugieren que las células que tienen activada la telomerasa son seleccionadas para continuar creciendo en cultivo. Estas células tienen una ventaja de crecimiento respecto a las células que no tienen activada la telomerasa, que mueren. Se ha sugerido que esta activación de la telomerasa contribuye a la formación de tumores in vivo.
Evidencias acumuladas a lo largo de los ańos sugieren que los modelos de cultivo de tejidos no reflejan el camino por el cual la telomerasa es activada en tumores in vivo. La telomerasa está activada en muchos tipos de cánceres humanos. Como la formación del tumor es un proceso de múltiples etapas que requiere un número determinado de cambios en la célula, la inmortalización celular in vitro sólo es un paso a lo largo del camino para la formación de un tumor. Sin embargo, hay evidencias de que el comportamiento del telómero y de la telomerasa en tumores no mimetiza el modelo de cultivo de tejidos. La longitud del telómero a menudo no está correlacionada con el estadio del tumor y la telomerasa es a menudo activada durante la ausencia del acortamiento de los telómeros. Además, la viabilidad de la telomerasa de un ratón knock-out durante seis generaciones apoya el argumento de que la activación de la telomerasa en tumores de ratón ocurre antes de que se alcance el acortamiento crítico de los telómeros. En tumores de ratón, la activación de la telomerasa ocurre en células que han pasado 30-40 divisiones celulares. El echo de que la telomerasa, de seis generaciones, que proviene de ratones knock-out derive de células que han pasado por más de 300 divisiones celulares, sugiere que el acortamiento crítico de los telómeros no ocurre hasta que al menos se llegue hasta esas divisiones celulares. Estos experimentos sugieren que in vivo, en tumores de ratón, la telomerasa es activada directamente o como consecuencia de un cambio regulatorio global, en vez de por la selección de células que pasaron a través de un estadio de acortamiento crítico del telómero y que tienen activada la telomerasa de manera estocástica. Estudios recientes basados en la regulación de la subunidad catalítica de la telomerasa apoyan los trabajos para determinar la regulación de la actividad telomerásica.
La telomerasa es activada por la oncoproteína viral HPV E6 (human pappillomavirus E6) y por el oncogen celular MYC (también conocido como c-MYC). Además, estudios recientes muestran que la oncoproteína viral HPV E7 (human pappillomavirus E7) también activa la telomerasa. La expresión de HPV E6 y E7 conjuntamente es otra vía para generar variantes inmortales. Finalmente, se sabe que el oncogen MYC regula la subunidad catalítica de la telomerasa, TERT, y esta TERT coopera con el oncogen viral HPV E7 en la inmortalización de las células.
MECANISMOS GENÉTICOS DEL CÁNCER: ONCOGENES Y GENES SUPRESORESDE TUMORES
Se amite que al menos un millar de genes funcionan de forma anómala en una célula cancerosa con malignidad máxima. Las alteraciones de estos genes son de diversa índole: mutaciones, amplificaciones, genes silenciosos... que tienden a acumularse.
El cáncer es el resultado de una serie de accidentes genéticos al azar sujetos a selección natural, por lo que no es probable que se produzcan dos casos de cáncer genéticamente idénticos, incluso aunque sean de la misma enfermedad.
Los genes reguladores normales pueden clasificarse en aquellos cuyos productos estimulan la división celular y aquellos cuyos productos la inhiben. Por lo tanto, existen dos caminos de mutación hacia la proliferación celular incontrolada y la invasividad. El primero consiste en que un gen estimulador se vuelva hiperactivo; este tipo de mutación tiene efecto dominante. El gen alterado se denomina oncogén y su alelo normal proto-oncogén. El segundo camino consiste en inactivar un gen inhibidor, cuyo efecto mutacional es recesivo y el gen implicado se denomina gen supresor de tumores.
El estudio genético de las células cancerosas se puede realizar de diversas maneras y generalmente las distintas vías conducen a la identificación de los mismos oncogenes. Un proto-oncogén puede identificarse a través de su presencia en retrovirus transformantes (esta cuestión es analizada con más detalle en el apartado titulado "virus y cáncer"), además de por las mutaciones que causan las inserciones de los genomas retrovíricos, la amplificación y transfección de genes en células cancerosas. También pueden buscarse más directamente, introduciendo secuencias de DNA de células cancerosas humanas que puedan producir proliferación incontrolada en células no cancerosas. Asismismo, es posible estudiar el cariotipo de las células tumorales, cuyos cromosomas suelen mostrar patrones concretos de translocación para tipos tumorales concretos. A partir de estudios de secuenciación del DNA, parece haberse determinado que en algunos casos la translocación podría transformar un proto-oncogén en un oncogén por fusión del primero con otro gen, resultando en una producción de proteína anormal; en otros casos la translocación trasladaría al proto-oncogén a un entorno cromosómico inapropiado que modificaría su transcripción, produciendo cantidades excesivas de la proteína para la que codifica.
La mayoría de proto-oncogenes codifican componentes de mecanismos reguladores de las seńales intercelulares que empujan a las células hacia la proliferación, la diferenciación y la apoptosis. Sus productos pertenecen a todos los tipos de moléculas que participan en la seńalización celular: proteínas secretadas, de unión a GTP, quinasas, de regulación génica, receptores transmembrana... las cuales realizan sus función en reacciones en cadena. Es comprensible que su alteración cause una proliferación descontrolada, debido a que una mutación en los genes que las codifican puede alterarlas de tal modo que dejen de hacer su función o transmitan seńales incorrectas, amplificadas o a destiempo.
El estudio de tumores humanos ha revelado que en realidad las células cancerosas tienen un elevado número de mutaciones. El genoma de las células cancerosas parece ser muy inestable, lo que causa una altísima frecuencia de mutaciones, que en muchas ocasiones no puede explicarse en base a la velocidad de aparición de mutaciones espontáneas durante la replicación del DNA. Como resultado de este descubrimiento, se ha propuesto que las células cancerosas presentan un fenotipo mutador (también llamado RER o "Replication Error"), es decir, que han adquirido una capacidad anormalmente alta de mutar sus genes. A favor de esta hipótesis, se ha demostrado una gran inestabilidad de microsatélites (secuencias cortas repetidas que se localizan en sitios relativamente constantes del genoma) en células tumorales humanas. Aunque los microsatélites no contienen genes codificantes y no afectan el fenotipo celular, sus alteraciones sí son por definición mutaciones, e implican que los genes en los que se encuentran están sufriendo alteraciones. El fenotipo mutador se asocia en cánceres colo-rectales y a otros que afectan al aparato digestivo preferentemente.
Se ha observado que la inestabilidad de microsatélites en células de cánceres humanos se asocia a alteraciones en ciertos genes que son homólogos a los que en microorganismos codifican por las proteínas que reparan los errores que tienen lugar durante la replicación del DNA por formación de pares de bases erróneos. Son los genes de reparación de los errores de replicación del DNA o "MisMatch Repair genes" o genes MMR. Se ha calculado que la mutación de estos genes causa un aumento de cien a mil veces en la acumulación de mutaciones en las células. Está comprobado especialmente en el cáncer colo-rectal hereditario de tipo no-poliposo (HNPCC, del inglés "Hereditary NonPolyposis Colon Cancer"), habiéndose descubierto hasta ahora seis genes implicados en la reparación del DNA en el hombre cuya alteración se asocia a la aparición de cáncer: MSH2, MLH1, PMS1, PMS2, GTBP, MSH3.
Un sistema distinto se encarga de la reparación de las lesiones en el DNA causadas por la luz ultravioleta o carcinógenos químicos: por ejemplo, de los dímeros de timina inducidos por la luz ultravioleta, de los complejos formados por la unión covalente de agentes químicos (como los benzopirenos del tabaco a residuos de guanina, G, del DNA) o de la eliminación de las bases que son metiladas por agentes alquilantes como los empleados en quimioterapia anticancerosa. Para la reparación de estas alteraciones de mayor consideración existe un complejo mecanismo denominado reparación por excisión de nucleótidos ("Nucleotide Excision Repair", NER, o "excinucleasa") compuesto en el hombre por al menos 17 proteínas. Algunos genes NER están alterados en tres síndromes hereditarios que conllevan una elevada frecuencia de cánceres: Xeroderma pigmentosum, síndrome de Cockayne y tricotiodistrofia.
Se pensaba que la elevada tasa de mutación observada en los tumores era debida al azar, pero al estudiarse detenidamente se descubrió que las mutaciones nunca son neutras, es decir, siempre inducen la activación y/o sobreexpresión de genes que van a favorecer al tumor y nunca a perjudicarlo.
Hay que tener en cuenta que además de la especial función de los oncogenes en el proceso de cancerización, la deficiencia concreta de las enzimas que se encargan de conservar y reparar el genoma es igualmente un punto favorecedor del cáncer. Aunque en su caso no se trata de una inducción directa, sino de una fragilización del genoma que conduciría a una mayor tasa de mutación y, por tanto, una mayor probabilidad de que un oncogén sea activado o bien un gen supresor de tumores se vea inhibido.
CONVERSIÓN DE UN PROTO-ONCOGÉN EN UN ONCOGÉN
Esta conversión puede darse de diferentes maneras:
Y el cambio puede darse de diversas maneras:
Cada gen particular y cada respuesta particular a los agentes carcinógenos presenta tipos característicos de anomalías.
Sin embargo, no hay que olvidar que el cáncer es el resultado de múltiples mutaciones y que el hecho de presentar a los oncogenes como causantes de transformación de una manera dominante y relativamente individual es debido a que los ensayos de laboratorio utilizan típicamente líneas celulares ya de por sí muy mutadas y, por ello, muy susceptibles a ser transformadas por un solo cambio genético particular. En realidad, en un organismo un solo oncogén no es capaz de convertir una célula normal en cancerosa.
La acción sinérgica de dos o más oncogenes específicos para generar células cancerígenas se conoce como colaboración oncogénica y se manifiesta tanto "in vitro" como "in vivo". Esta colaboración sería necesaria debido a que en la regulación de la proliferación celular actúan varios mecanismos en paralelo, así el error de un solo componente no es suficiente para causar el desajuste celular.